|
|
Post by Admin on Jun 4, 2020 7:24:49 GMT
 Fig. 3 Structure analysis of the RBM and ACE2 interface. (A) SARS-CoV and SARS-CoV-2 receptor binding domains (RBD). Human ACE2 in green (PDB 6M0J) at the top and the RBD of the S-protein at the bottom; SARS-CoV S-protein (PDB 2AJF) in red, and SARS-CoV-2 S-protein (PDB 6M0J) in magenta with RBM in blue. All structure backbones shown as ribbons with key residues at the interface shown as stick models, labeled using the same color scheme. (B) Impact of different RBM amino acids between SARS-CoV-2 RaTG13 on ACE2 binding. (C) Impact of an amino acid at position 498 (Q in SARS-CoV-2, top, and H in RaTG13, bottom) on ACE2 binding. Same color-coding as in (A) with additional hydrogen bonding as light blue lines. (D) Impact of two deletions on ACE2 binding interface in some bat-SL-CoVs, positions indicated in yellow, and modeled structure with long deletion between residue 473 in light blue. Three small insertions are identical in SARS-CoV-2 and RaTG13 but not found in other CoVs in the Sarbecovirus group (27, 28). The RaTG13 sequence was sampled in 2013, years before SARS-CoV-2 was first identified. It is unlikely that both SARS-CoV-2 and RaTG13 independently acquired identical insertions at three different locations in the S gene. Thus, it is plausible that a RaTG13-like virus served as a progenitor to generate SARS-CoV-2 by gaining a complete human ACE2 binding RBM from Pan_SL-CoV_GD-like viruses through recombination. Genetic divergence at the nucleic acid level between Wuhan-Hu-1 and Pan_SL-CoV_GD viruses is significantly reduced from 13.9% (Fig. 1E) to 1.4% at the amino acid level (Fig. 2B) in the RBM region, indicating recombination between RaTG13-like CoVs and Pan_SL-CoV_GD-like CoVs. Furthermore, SARS-CoV-2 has a unique furin cleavage site insertion (PRRA) not found in any other CoVs in the Sarbecovirus group (Fig. S3) (27), although similar motifs are also found in MERS and more divergent bat CoVs (29). This PRRA motif makes the S1/S2 cleavage in SARS-CoV-2 much more efficient than in SARS-CoV and may expand its tropism and/or enhance its transmissibility (20). A recent study of bat CoVs in Yunnan, China, identified a three-amino acid insertion (PAA) at the same site (30). Although it is not known if this PAA motif can function like the PRRA motif, the presence of a similar insertion at the same site indicates that such insertion may already be present in the wild bat CoVs. The more efficient cleavage of S1 and S2 subunits of the spike glycoprotein (29) and efficient binding to ACE2 by SARS-CoV-2 (20, 25) may have allowed SARS-CoV-2 to jump to humans, leading to the rapid spread of SARS-CoV-2 in China and the rest of the world. Strong purifying selection among SARS-CoV-2 and closely related viruses Recombination from Pan_SL-CoV_GD at the RBM and at the unique furin cleavage site insertion prompted us to examine the SARS-CoV-2 sequences within these regions. Amino acid sequences from SARS-CoV-2, RaTG13, and all Pan_SL-CoV viruses (group A) are identical or nearly identical in the region before and after the RBM and at the region after the furin cleavage site (S2 subunit), while all other CoVs (group B) are very distinctive (Fig. 4A and S4). The average of all pairwise dN/dS ratios, defined as ω, among SARS-CoV-2, RaTG13, and Pan_SL-CoV viruses at the S2 subunit is ω = 0.013, compared to the much higher values ω =0.053 in the S1 region preceding the furin cleavage site, and ω = 0.042 at the S2 subunit for all other CoVs (Fig. 4B). The much lower ω value at the S2 subunit among the SARS-CoV-2, RaTG13, and Pan_SL-CoV viruses indicates that this region is under strong purifying selection within these sequences. A plot of synonymous and nonsynonymous substitutions relative to Wuhan-Hu-1 highlights the regional differences across the region before and after the furin cleavage site (Fig. 4A): the S2 subunit is highly conserved among the SARS-CoV-2, RaTG13, and Pan_SL-CoV viruses (group A), while far more nonsynonymous mutations are observed in the rest of the CoV sequences (group B). The shift in selective pressure at the S1/S2 cleaveage site among these related viruses versus other CoVs begins near codon 368 (Fig. 4B): the two graphs show the cumulative plots of the average behavior of each codon for all pairwise comparisons in the input data, for synonymous mutations, non-synonymous mutations and indels of group A sequences and group B sequences. The non-synonymous plot shows a marked change in slope (vertical step) in the group A sequences at codon 368, but not in group B sequences. Similarly, when looking at all the dS/dN ratios (ω) for each group A sequence compared to the Wuhan-Hu-1 sequence, we see that these ratios are much lower in the 5′-end of the region, before codon 368 (nucleic acid position 1104), compared to the 3′-end, and no such difference is observed in the group B sequences (Fig. 4C).  Fig. 4 Strong purifying selection after furin cleavage in S gene among SARS-CoV-2 and closely related viruses. (A) Phylogenetic tree (left) and Highlighter plot (right) of sequences around the RBM and furin cleavage site compared to SARS-CoV-2 Wuhan-Hu-1 (na positions 22541-24391). ACE2 receptor binding motif (RBM) and furin cleavage site highlighted in light-gray boxes. Mutations compared to Wuahn-Hu-1 are light blue for synonymous, red for non-synonymous. Dominance of synonymous mutations within group A compared to group B highlighted on the right. (B) Cumulative plots of each codon average behavior for all pairwise comparisons for indels and synonymous (light blue) and non-synonymous (red) mutations, by group. The abrupt slope change of the nonsynonymous curve in group A at around codon 368 (na 1104) is indicative of a shift in localized accumulations of non-synonymous mutations after the furin cleavage site. Group B instead lacks this abrupt change in slope at the same position. Values of ω denote average ratios of the rate of nonsynonymous substitutions per nonsynonymous site (dN/dS) for each group and region. (C) Sequence dS/dN ratios compared to Wuhan-Hu-1 within codons 1-368 (na 1-1104, green) and codons 369-620 (na 1105-1893, dark blue) in group A and group B sequences. This strong purifying selection observed in the S2 subunit of the S gene is not surprising given its role in cell entry by fusing the viral and host cell membranes (5, 19). Following the binding of RBD to the ACE2 receptor, heptad repeat regions 1 (HR1) and 2 (HR2) within the S2 subunit rearrange to form the fusion core, bringing together the viral and cell membranes for fusion and infection (Fig. S5A). Due to the mechanistic constraints for this assembly for fusion, the protein segments that take part in this assembly are well preserved (20, 31). Furthermore, some regions of the S2 subunit are covered by S1 in the trimer conformation of the spike protein (Fig. S5B). Based on the currently available, but incomplete, cryo-EM structure of the spike trimer, we estimate that 60%-65% of S2 amino acids are buried. This adds further structural constraints on changing amino acids in S2. While hundreds of new SARS-CoV-2 sequences are added to the GISAID repertoire every day (32), we note that the RBM region currently remains highly conserved. No amino acid within 6 Angstroms of the ACE2 binding site has repeated variations, with the exception of G476S, a very rare mutation found in 8 sequences from a local cluster in Washington state, out of 6,400 total sequences from GISAID (April 13, 2020). In addition, we observe similar patterns of purifying selection pressure in other parts of the genome, including the E and M genes, as well as the partial ORF1a and ORF1b genes (Fig. S6 and S7). Interestingly, the viruses affected by purifying selection pressure varies depending on which genes are analyzed. SARS-CoV-2, RaTG13, all Pan_SL-CoV and the two bat CoVs (ZXC21 and ZC45) are under the similar purifying selection in both the E and M genes (Figs. 5A and S6). In the S2 subunit, similar purifying selection are only observed for SARS-CoV-2, RaTG13, and all Pan_SL-CoV (Fig. 5B). A few viruses including only SARS-CoV-2, RaTG13, and pangolin CoVs from Guangdong are under similar purifying selection in the partial regions of ORF1a and ORF1b (Figs. 5C and S7). Strong purifying selection pressure on SARS-CoV-2, RaTG13 and Pan_SL-CoV_GD viruses, as indicated by consistently low ω values, suggests that these complete and partial genes are under similar functional/structural constraints among the different host species. In two extreme cases, amino acid sequences of the E gene and the 3′ end of ORF1a are identical among the compared CoV sequences, although genetic distances are quite large among these viruses at the nucleic acid level (Fig. 5A and 5C). Such evolutionary constraints in many parts of the viral genome, especially at functional domains in the S gene which plays an important role in cross-species transmission (5, 12), coupled with frequent recombination, may facilitate cross-species transmissions between RaTG13-like bat and/or Pan_SL-CoV_GD-like viruses.  Fig. 5 Strong purifying selection on complete and partial gene regions among SARS-CoV-2, RaTG13 and Pan_SL-CoV viruses. Purifying selection pressure on complete and partial genes among different viruses (red boxes) as evident by shorter branches in amino acid trees compared to nucleic acid trees. Distinct purifying selection patterns are observed among different viruses: (A) SARS-CoV-2, RaTG13, all Pan_SL-CoV and bat CoV ZXC21 and ZC45; (B) SARS-CoV-2, RaTG13, all Pan_SL-CoV sequences; (C) SARS-CoV-2, RaTG13 and Pan_SL-CoV_GD. Cumulative plots of the average behavior of each codon for all pairwise comparisons for synonymous mutations, non-synonymous mutations and indels within each gene region. ω denotes the average ratio of the rate of nonsynonymous substitutions per nonsynonymous site (dN/dS) for each group. |
|
|
|
Post by Admin on Jun 5, 2020 7:27:25 GMT
Frequent recombination between SARS-CoVs and bat_SL-CoVs Previous studies using more limited sequence sets found that SARS-CoVs originated through multiple recombination events between different bat-CoVs (10, 12, 21, 33, 34). Our phylogenetic analyses of individual genes confirm this and show that SARS-CoV sequences tend to cluster with YN2018B, Rs9401, Rs7327, WIV16 and Rs4231 (group A) for some genes and Rf4092, YN2013, Anlong-112 and GX2013 (group B) for others (Fig. S8). SimPlot analysis using both groups of bat_SL-CoVs and the closely related bat CoV YNLF-34C (34) shows that SARS-CoV GZ02 shifts in similarity among different bat SL-CoVs at various regions of the genome (Fig. 6A). In particular, phylogenetic reconstruction of the beginning of ORF1a (region 1) confirms that SARS-CoVs cluster with YNLF-34C (34), and this cluster is distinctive comparing to all other CoVs (Fig. 6B). YNLF-34C is more divergent from SARS-CoV than other bat-CoV viruses before and after this region, confirming the previously reported complex recombinant nature of YNLF-34C (34) (Fig. 5A). At the end of the S gene (region 2), SARS-CoVs cluster with group A CoVs, forming a highly divergent clade (Fig. 6C). In region 3 (ORF8), SARS-CoVs and group B CoVs, together with YNLF-34C, form a very divergent and distinctive cluster (Fig. 6D). To further explore the recombinant nature of SARS-CoVs, we compared GZ02 to representative bat CoV sequences using the RIP recombination detection tool (18). We identified four significant breakpoints (at 99% confidence) between the two parental lineages (Fig. S9A), further supported by phylogenetic analysis (Fig. S9B-S9D). In addition, the two aforementioned groups of bat CoVs (shown in light brown and light blue in the trees) show similar cluster changes across the five recombinant regions, suggesting multiple events of historic recombination among bat SL-CoVs. These results demonstrate that SARS-CoV shares a recombinant history with at least three different groups of bat-CoVs and confirms the major role of recombination in the evolution of these viruses.  Fig. 6 Multiple recombination of SARS-CoVs with different bat_SL-CoVs. (A) SimPlot genetic similarity plot between SARS-CoV GZ02 and SARS_SL-CoVs, using a 400-bp window at a 50-bp step and the Kimura 2-parameter model. Group A CoVs (YN2018B, Rs9401, Rs7327, WIV16 and Rs4231) are shown in blue, group B CoVs (Rf4092, YN2013, Anlong-112 and GX2013) in orange, YNLF-34C in green, and outlier control HKU3-12 in red. Phylogenetic trees for high similarity regions between GZ02 and YNLF-34C (B), group A (C), and group B (D). All positions are relative to Wuhan-Hu-1. Of the bat SL-CoVs that contributed to the recombinant origin of SARS-CoV, only group A viruses bind to ACE2. Group B bat SL-CoVs do not infect human cells (5, 21, 22) and have two deletions in the RBM (Figs. 1E and 2A). The short deletion between residues 445 and 449, and in particular the loss of Y449, which forms three hydrogen bonds with ACE2, will significantly affect the overall structure of the RBM (Figs. 3C and 3D). The region encompassing the large deletion between residues 473 and 486 contains the loop structure that accounts for the major differences between the S protein of SARS-CoV and SARS-CoV-2 (Fig. 3A) and strengthen the interaction of the latter to ACE2 (26). This deletion causes the loss of contact site F486 and affects the conserved residue F498’s hydrophobic interaction with residue M82 on ACE2 (Fig. 3D). These two deletions will render RBM in those CoVs incapable of binding human ACE2. Therefore, recombination may play a role in enabling cross-species transmission in SARS-CoVs through the acquisition of an S gene type that can efficiently bind to the human ACE2 receptor. ORF8 is one of the highly variable genes in coronaviruses and its function has not yet been well elucidated (5, 12, 35). Recombination breakpoints within this region show that recombination occurred at the beginning and the end of ORF8 (Fig. S10), where nucleic acid sequences are nearly identical among both SARS-CoVs and group B bat CoVs. Moreover, all compared viruses form three highly distinct clusters (Fig. 6D), suggesting that the ORF8 gene may be biologically constrained and evolves through modular recombination. The third recombination region at the beginning of ORF1a is near where SARS-CoV-2 also recombined with other bat CoVs (region 1 in Fig. 1A). This region is highly variable (5, 12) and recombination within this part of the genome was also found in other CoVs, suggesting that it may be a recombination hotspot and may factor into cross-species transmission. Discussion There are three important aspects to betacoronavirus evolution that should be carefully considered in phylogenetic reconstructions among more distant coronaviruses. First, there is extensive recombination among all of these viruses (10, 12, 21, 33, 34) (Figs. 1 and 5), making standard phylogenetic reconstructions based on full genomes problematic, as different regions of the genome have distinct ancestral relationships. Second, between more distant sequences, synonymous substitutions are often fully saturated, which can confound analyses of selective pressure and adds noise to phylogenetic analysis. Finally, there are different selective pressures at work in different lineages, which is worth consideration when interpreting trees. The currently sampled pangolin CoVs are too divergent from SARS-CoV-2 to be its recent progenitors, but it is noteworthy that these sequences contain an RBM that can most likely bind to human ACE2. While RaTG13 is the most closely related CoV sequence to SARS-CoV-2, it has a distinctive RBM. In addition, a recent study showed that the RaTG13 pseudovirus is much less efficient than the SARS-CoV-2 pseudovirus in using ACE2 to infect cells (26). SARS-CoV-2 has a nearly identical RBM to the one found in the pangolin CoVs from Guangdong. Thus, it is plausible that RaTG13-like bat-CoV viruses may have obtained the RBM sequence binding to human ACE2 through recombination with Pan_SL-CoV_GD-like viruses. We hypothesize that this, and/or other ancestral recombination events between viruses infecting bats and pangolins, may have played a key role in the evolution of the strain that lead to the introduction of SARS-CoV-2 into humans. It is also possible that other not yet identified hosts infected with CoVs that can jump to human populations through cross-species transmission if they can successfully infect human cells through ACE2 or other receptors. Interestingly, an analysis of 6,400 SARS-CoV-2 sequences from GISAID (Global Initiative on Sharing All Influenza Data) (36, 37) identifies only one very rare mutation, G476S that is directly in a ACE2 contact residue. It was found in a local cluster of sequences from Washington state. However, it is at the periphery of the receptor contact surface, and so may not significantly impact the virus’s receptor binding affinity. All three human CoVs (SARS, MERS and SARS-2) are the result of recombination among CoVs. Recombination in all three viruses involved the S gene, likely a precondition to zoonosis that enabled efficient binding to human receptors (5, 12). Extensive recombination among bat coronaviruses and strong purifying selection pressure among viruses from humans, bats and pangolins may allow such closely related viruses to readily jump between species and adapt to the new hosts. Many bat CoVs have been found able to bind to human ACE2 and replicate in human cells (10, 21, 22, 38–40). Serological evidence has revealed that additional otherwise undetected spillovers have occurred in people in China living in proximity to wild bat populations (41). Continuous surveillance of coronaviruses in their natural hosts and in humans will be key to rapid control of new coronavirus outbreaks. While the SARS and MERS originating strains have been found in civets and dromedary camels respectively (14, 15), so far, efforts to identify a similarly close link in the original pathway of SARS-CoV-2 into humans have failed. If the new SARS-CoV-2 strain did not cause widespread infections in its natural or intermediate hosts, such a strain may never be identified. The close proximity of animals of different species in a wet market setting may increase the potential for cross-species spillover infections, by enabling recombination between more distant coronaviruses and the emergence of recombinants with novel phenotypes. While the direct reservoir of SARS-CoV-2 is still being sought, one thing is clear: reducing or eliminating direct human contact with wild animals is critical to preventing new coronavirus zoonosis in the future. |
|
|
|
Post by Admin on Oct 6, 2020 6:45:45 GMT
Numerous animals may be vulnerable to SARS-CoV-2, the virus that causes COVID-19, according to a large study modelling how the virus might infect different animals' cells, led by UCL researchers. The study, published in Scientific Reports, reports evidence that 26 animals regularly in contact with people may be susceptible to infection. The researchers investigated how the spike protein from SARS-CoV-2 could interact with the ACE2 protein it attaches to when it infects people. The focus of the investigation was whether mutations in the ACE2 protein in 215 different animals, that make it different from the human version, would reduce the stability of the binding complex between the virus protein and host protein. Binding to the protein enables the virus to gain entry into host cells; while it is possible the virus might be able to infect animals via another pathway, it is unlikely based on current evidence that the virus could infect an animal if it cannot form a stable binding complex with ACE2.  The researchers found that for some animals, such as sheep and great apes (chimpanzee, gorilla, orangutan, and bonobo, many of which are endangered in the wild), the proteins would be able to bind together just as strongly as they do when the virus infects people. Some of the animals, such as sheep, have not yet been studied with infection tests, so this does not confirm that the animal can indeed be infected. Lead author Professor Christine Orengo (UCL Structural & Molecular Biology) said: "We wanted to look beyond just the animals that had been studied experimentally, to see which animals might be at risk of infection, and would warrant further investigation and possible monitoring. "The animals we identified may be at risk of outbreaks that could threaten endangered species or harm the livelihoods of farmers. The animals might also act as reservoirs of the virus, with the potential to re-infect humans later on, as has been documented on mink farms." The research team also performed more detailed structural analyses for certain animals, to gain a better understanding of how infection risks may differ across animal species. By comparing their findings to other experimental data, they set thresholds to predict which animals are at risk of infection, and which ones most likely cannot be infected. They found that most birds, fish, and reptiles do not appear to be at risk of infection, but the majority of the mammals they reviewed could potentially be infected. Professor Orengo added: "The details of host infection and severity of response are more complex than just the interactions of the spike protein with ACE2, so our research is continuing to explore interactions involving other host virus proteins." The team's findings mostly agree with experiments conducted in living animals and with reported cases of infections. They predict possible infection in domestic cats, dogs, mink, lions, and tigers, all of which have had reported cases, as well as ferrets and macaques, which have been infected in laboratory studies.  First author, Su Datt Lam (UCL Structural & Molecular Biology and the National University of Malaysia) said: "Unlike laboratory-based experiments, the computational analyses we devised can be run automatically and rapidly. Therefore, these methods could be applied easily to future virus outbreaks that, unfortunately, are becoming more common due to human encroachment into natural habitats." Co-author Professor Joanne Santini (UCL Structural & Molecular Biology) said: "To protect animals, as well as to protect ourselves from the risk of one day catching COVID-19 from an infected animal, we need large-scale surveillance of animals, particularly pets and farm animals, to catch cases or clusters early on while they're still manageable. "It may also be important to employ hygiene measures when dealing with animals, similar to the behaviours we've all been learning this year to reduce transmission, and for infected people to isolate from animals as well as from other people." Explore further SARS-CoV-2 transmission to animals: Monitoring needed to mitigate risk More information: Scientific Reports (2020). DOI: 10.1038/s41598-020-71936-5 |
|
|
|
Post by Admin on Oct 6, 2020 21:44:28 GMT
SARS-CoV-2 spike protein predicted to form complexes with host receptor protein orthologues from a broad range of mammals
S. D. Lam, N. Bordin, V. P. Waman, H. M. Scholes, P. Ashford, N. Sen, L. van Dorp, C. Rauer, N. L. Dawson, C. S. M. Pang, M. Abbasian, I. Sillitoe, S. J. L. Edwards, F. Fraternali, J. G. Lees, J. M. Santini & C. A. Orengo
Scientific Reports volume 10, Article number: 16471 (2020)
Abstract
SARS-CoV-2 has a zoonotic origin and was transmitted to humans via an undetermined intermediate host, leading to infections in humans and other mammals. To enter host cells, the viral spike protein (S-protein) binds to its receptor, ACE2, and is then processed by TMPRSS2. Whilst receptor binding contributes to the viral host range, S-protein:ACE2 complexes from other animals have not been investigated widely. To predict infection risks, we modelled S-protein:ACE2 complexes from 215 vertebrate species, calculated changes in the energy of the complex caused by mutations in each species, relative to human ACE2, and correlated these changes with COVID-19 infection data. We also analysed structural interactions to better understand the key residues contributing to affinity. We predict that mutations are more detrimental in ACE2 than TMPRSS2. Finally, we demonstrate phylogenetically that human SARS-CoV-2 strains have been isolated in animals. Our results suggest that SARS-CoV-2 can infect a broad range of mammals, but few fish, birds or reptiles. Susceptible animals could serve as reservoirs of the virus, necessitating careful ongoing animal management and surveillance.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel coronavirus that emerged towards the end of 2019 and is responsible for the coronavirus disease 2019 (COVID-19) global pandemic. Available data suggests that SARS-CoV-2 has a zoonotic source1, with the closest sequence currently available deriving from the horseshoe bat2. As yet, the transmission route to humans, including the intermediate host, is unknown. So far, little work has been done to assess the animal reservoirs of SARS-CoV-2, or the potential for the virus to spread to other species living with, or in close proximity to, humans in domestic, rural, agricultural or zoological settings.
Coronaviruses, including SARS-CoV-2, are major multi-host pathogens and can infect a wide range of non-human animals3,4,5. SARS-CoV-2 is a member of the Betacoronavirus genus, which includes viruses that infect economically important livestock, including cows6 and pigs7, together with mice8, rats9, rabbits 10, and wildlife, such as antelope and giraffe11. Severe acute respiratory syndrome coronavirus (SARS-CoV), the betacoronavirus that caused the 2002–2004 SARS outbreak12, likely jumped to humans from its original bat host via civets. Viruses genetically similar to human SARS-CoV have been isolated from animals as diverse as raccoon dogs, ferret-badgers3 and pigs13, suggesting the existence of a large host reservoir. It is therefore probable that SARS-CoV-2 can also infect a wide range of species.
Real-world SARS-CoV-2 infections have been reported in cats14, lions and tigers15, dogs16,17 and minks16,17. Animal infection studies have also identified cats18 and dogs18 as hosts, as well as ferrets18, macaques19 and marmosets19. Recent in vitro studies have also suggested an even broader set of animals may be infected20,21,−22. To understand the potential host range of SARS-CoV-2, the plausible extent of zoonotic and anthroponotic transmission, and to guide surveillance efforts, it is vital to know which species are susceptible to SARS-CoV-2 infection.
The receptor binding domain (RBD) of the SARS-CoV-2 spike protein (S-protein) binds to the extracellular peptidase domain of angiotensin I converting enzyme 2 (ACE2) mediating cell entry23. The sequence of ACE2 is highly conserved across vertebrates, suggesting that SARS-CoV-2 could use orthologues of ACE2 for cell entry. The structure of the SARS-CoV-2 S-protein RBD has been solved in complex with human ACE224. Identification of critical binding residues in this structure have provided valuable insights into viral recognition of the host receptor24,25,26,27,28,29. Deep mutagenesis studies have also revealed residues important for stability30,31. Compared with SARS-CoV, the SARS-CoV-2 S-protein has a 10–22-fold higher affinity for human ACE224,25,32, due to more contacts in the interface that cover a larger surface area29, and three mutational hotspots in the S-protein that lead to a more specific and compact conformation29,33. Similarly, variations in human ACE2 have also been found to increase affinity for S-protein receptor binding34. These factors may contribute to the host range and infectivity of SARS-CoV-2.
Both SARS-CoV-2 and SARS-CoV additionally require the transmembrane serine protease (TMPRSS2) to mediate cell entry. Together, ACE2 and TMPRSS2 confer specificity of host cell types that the virus can enter35,36. Upon binding to ACE2, the S-protein is cleaved by TMPRSS2 at two cleavage sites on separate loops, which primes the S-protein for cell entry37. TMPRSS2 has been docked against the SARS-CoV-2 S-protein, which revealed its binding site to be adjacent to these two cleavage sites36. An approved TMPRSS2 protease inhibitor drug is able to block SARS-CoV-2 cell entry38, which demonstrates the key role of TMPRSS2 alongside ACE239. As such, both ACE2 and TMPRSS2 represent attractive therapeutic targets against SARS-CoV-240.
Recent work has predicted possible hosts for SARS-CoV-2 using the structural interplay between the S-protein and ACE2. These studies proposed a broad range of hosts, covering hundreds of mammalian species, including tens of bat 27 and primate 41 species, and more comprehensive studies analysing all classes of vertebrates 41,42,43, including agricultural species of cow, sheep, goat, bison and water buffalo. In addition, sites in ACE2 have been identified as under positive selection in bats, particularly in regions involved in binding the S-protein27,44. The impacts of mutations in ACE2 orthologues have also been tested, for example structural modelling of ACE2 from 27 primate species41 demonstrated that apes and African and Asian monkeys may also be susceptible to SARS-CoV-2. However, whilst cell entry is necessary for viral infection, it may not be sufficient alone to cause disease. For example, variations in other proteins may prevent downstream events that are required for viral replication in a new host. Hence, examples of real-world infections14,15,16,17 and experimental data from animal infection studies18,19,20,21,22 are required to validate hosts that are predicted to be susceptible.
Here, we analysed the effect of known mutations in orthologues of ACE2 and TMPRSS2 from a broad range of 215 vertebrate species, including primates, rodents and other placental mammals; birds; reptiles; and fish. For each species, we generated a 3-dimensional model of the ACE2 protein structure from its protein sequence and calculated the impacts of known mutations in ACE2 on the stability of the S-protein:ACE2 complex. We correlated changes in the energy of the complex with changes in the structure of ACE2, chemical properties of residues in the binding interface, and experimental COVID-19 infection phenotypes from in vivo and in vitro animal studies. To further test our predictions and rationalise the key sites contributing to energy changes of the complex, we performed detailed manual structural analyses, presented as a variety of case studies for different species. Unlike other studies that analyse interactions that the S-protein makes with the host, we also analysed the impact of mutations in vertebrate orthologues of TMPRSS2. Our results suggest that SARS-CoV-2 could infect a broad range of vertebrates, which could serve as reservoirs of the virus, supporting future anthroponotic and zoonotic transmission.
|
|
|
|
Post by Admin on Oct 7, 2020 5:38:50 GMT
Conservation of ACE2 in vertebrates We aligned protein sequences of 247 vertebrate orthologues of ACE2. Most orthologues have more than 60% sequence identity with human ACE2 (Supplementary Fig. 1A). For each orthologue, we generated a 3-dimensional model of the protein structure from its protein sequence using FunMod45,46. We were able to build high-quality models for 236 vertebrate orthologues, with nDOPE scores < − 1 (Supplementary Table 5); 11 low-quality models were removed from the analysis. 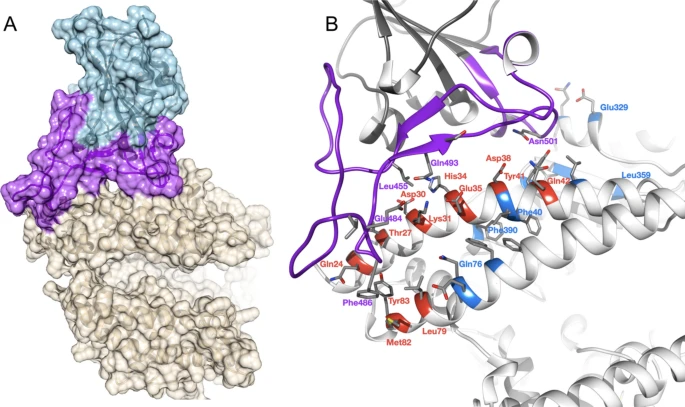 Figure 1 Overview of SARS-CoV-2 S-protein:human ACE2 complex and interface. (a) SARS-CoV-2 S-protein RBD (light blue, purple) showing the receptor binding motif (purple) at the interface with ACE2 (tan). (b) residues in the SARS-CoV-2 S-protein:human ACE2 complex interface. Key RBD interface residues are shown (purple) with a subset of ACE2 contact residues that are not conserved in vertebrates for DC (red) and DCEX residues (blue) (PDB ID 6M0J). Identification of critical S-protein:ACE2 interface residues ACE2 residues directly contacting the S-protein (DC residues) were identified in a structure of the complex (PDB ID 6M0J; Fig. 1a, Supplementary Results 2, Supplementary Fig. 3). We also identified an extended set of both DC residues and residues within 8 Å of DC residues likely to be influencing binding (DCEX residues). After analysing the orthologue interfaces, we removed models that were missing > 10 DCEX residues; 21 models were removed from the analysis, leaving 215 models to take forward for further analysis. We observed high sequence (> 60% identity) and structure similarity (score > 90 out of 100) between ACE2 proteins for all species (Supplementary Results 1). Changes in the energy of the S-protein:ACE2 complex in vertebrates We used multiple methods to assess the change in energy (ΔΔG, kcal/mol) of the SARS-CoV-2 S-protein:ACE2 complex following mutations in DC residues and DCEX residues that are likely to influence binding. We found that protocol 2 employing mCSM-PPI2 (henceforth referred to as P(2)-PPI2), calculated over the DCEX residues, correlated best with the phenotype data (Supplementary Results 3, Supplementary Fig. 4, Table 1), justifying the use of animal models to calculate ΔΔG values in this context. Since this protocol considers mutations from animal to human, lower ΔΔG values correspond to stabilisation of the animal complex relative to the human complex, and therefore higher risk of infection. We show the residues that P(2)-PPI2 reports as stabilising or destabilising for the SARS-CoV-2 S-protein:ACE2 animal complex for DC (Supplementary Fig. 8) and DCEX (Supplementary Fig. 9) residues. To consider ΔΔG values in an evolutionary context, we annotated phylogenetic trees for all 215 vertebrate species analysed (Supplementary Fig. 7) and for a subset of animals that humans come into close contact with in domestic, agricultural or zoological settings (Fig. 2). 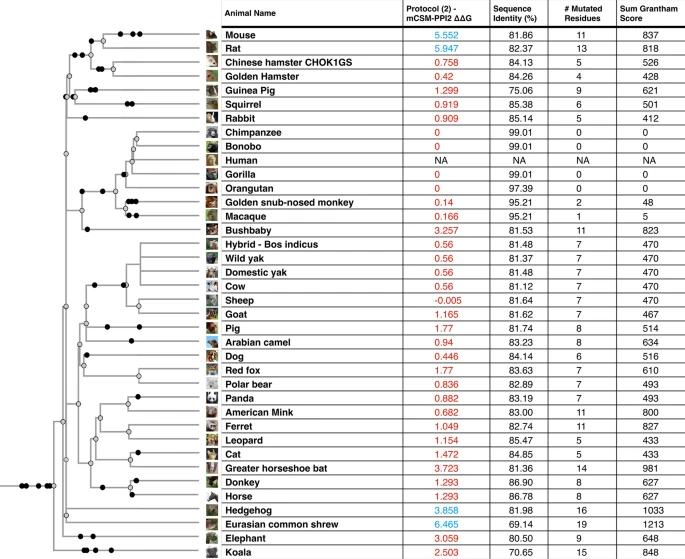 Figure 2 Phylogenetic tree of species that humans come into close contact with in domestic, agricultural or zoological settings. Leaves are annotated by the change in energy of the complex (ΔΔG), as measured by protocol 2 mCSM-PPI2. Animals are categorised according to risk of infection by SARS-CoV-2, with ΔΔG ≤ 3.7 being at risk (red), and ΔΔG > 3.7 not at risk (blue). These thresholds were chosen as they agree well with the available experimental data (Fig. 3). For each animal, the sequence identity to the human ACE2 sequence, the number of mutated residues compared to the human ACE2 sequence, and the total chemical shift (measured by Grantham score—see Supplementary Methods 5) across the DCEX residues are also shown. This tree contains a subset of animals from Supplementary Fig. 7. Animal photos courtesy of ENSEMBL and associated sources (https://www.ensembl.org/info/about/image_credits.html). |
|

