

Post by Admin on Sept 4, 2023 3:11:30 GMT
We investigated the contributions of genetic variants altering responses to SARS-CoV-2 ex vivo to COVID-19 risk in vivo by determining whether (r)eQTLs were more strongly associated with COVID-19 GWAS hits8 than random, matched SNPs (Methods). We observed an enrichment in eQTLs at loci associated with susceptibility (reported cases) and severity (hospitalized or critical cases) (FE = 4.1 and FE > 3.8, respectively, one-sided resampling, P < 10−4), and a specific enrichment in reQTLs at severity loci (FE > 3.7, one-sided resampling, P < 3 × 10−3; Fig. 5a). This trend was observed across most cell lineages (Extended Data Fig. 10a). Colocalization analyses identified 40 genes at which there was a high probability of (r)eQTL colocalization with COVID-19 hits (posterior probability that both traits are linked to the same SNP (PPH4 ) > 0.8) and transcriptome-wide association studies (TWASs) linked predicted gene expression with COVID-19 risk for 30 of these genes (FDRTWAS < 0.01; Supplementary Table 9a). These included direct regulators of innate immunity, such as IFNAR2 in non-stimulated CD4+ T cells, IRF1 in non-stimulated NK and CD8+ T cells, OAS1 in lymphoid cells stimulated with SARS-CoV-2 and IAV, and OAS3 in SARS-CoV-2-exposed CD16+ monocytes (Fig. 5b and Extended Data Fig. 10b,c). These results support a contribution of immunity-related (r)eQTLs to COVID-19 risk.
Fig. 5: eQTLs and reQTLs contribute to COVID-19 risk.
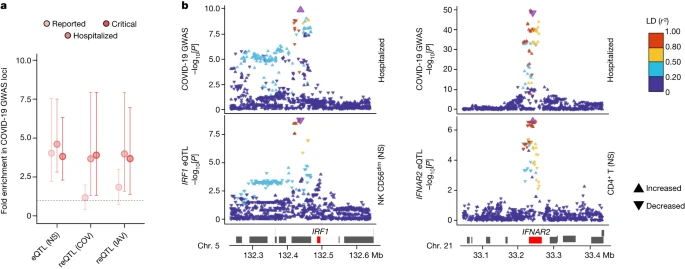
a, Enrichment in GWAS loci associated with COVID-19 susceptibility and severity at eQTLs and reQTLs. Data are the mean and 2.5th–97.5th percentiles (95% CIs) of fold enrichments observed over n = 10,000 resamplings. b, Colocalization of IRF1 and IFNAR2 eQTLs with COVID-19 severity loci. Top, the −log10
profiles (two-sided Student’s t-tests) for association with COVID-19-related hospitalization. Bottom, the −log10
profiles for association with expression in non-stimulated CD56dim NK cells (IRF1) and CD4+ T cells (IFNAR2). The colour code reflects the degree of LD (r2) with the consensus SNP identified by colocalization analyses (purple). For each SNP, the direction of the arrow indicates the direction of the effect. Chr., chromosome.
Focusing on the evolutionary factors affecting COVID-19 risk, we identified 20 eQTLs that (1) colocalized with COVID-19 hits (PPH4 > 0.8) and (2) presented positive selection signals (top 1% PBS, n = 13 eQTLs) or evidence of archaic introgression (n = 7 eQTLs), 14 of which regulate genes of which the expression is correlated with COVID-19 susceptibility and/or severity (FDRTWAS < 0.01) (Fig. 6). For example, two variants in high LD at DR1 (rs569414 and rs1559828, r2 > 0.73) displayed extremely high levels of population differentiation, probably due to selection outside Africa (DAF = 0.13 in Africa versus higher than 0.62 in Eurasia; Extended Data Fig. 10d). DR1 suppresses type I IFN responses49 and the selected alleles, which decrease COVID-19 severity, reduce DR1 expression in most immune cells (Fig. 6). Likewise, an approximately 39 kb Neanderthal haplotype, spanning the MUC20 locus in Eurasians, contains the rs2177336-T allele that increases MUC20 expression in SARS-CoV-2-stimulated cells, particularly for CD4+ T cells, and decreases COVID-19 susceptibility (Fig. 6). Together, these results reveal how past selection or Neanderthal introgression have impacted immune responses that contribute to present-day disparities in COVID-19 risk.