|
|
Post by Admin on Apr 23, 2018 18:28:14 GMT
The consensus view of the peopling of the Americas, incorporating archaeological, linguistic and genetic evidence, proposes colonization by a small founder population from Northeast Asia via Beringia 15–20 Kya (thousand years ago), followed by one or two additional migrations also via Alaska, contributing only to the gene pools of North Americans, and little subsequent migration into the Americas south of the Arctic Circle before the voyages from Europe initiated by Columbus in 1492 [1]. In the most detailed genetic analysis thus far, for example, Reich and colleagues [2] identified three sources of Native American ancestry: a ‘First American’ stream contributing to all Native populations, a second stream contributing only to Eskimo-Aleut-speaking Arctic populations, and a third stream contributing only to a Na-Dene-speaking North American population. Nevertheless, there is strong evidence for additional long-distance contacts between the Americas and other continents between these initial migrations and 1492. Norse explorers reached North America around 1000 CE and established a short-lived colony, documented in the Vinland Sagas and supported by archaeological excavations [3]. The sweet potato (Ipomoea batatas) was domesticated in South America (probably Peru), but combined genetic and historical analyses demonstrate that it was transported from South America to Polynesia before 1000–1100 CE [4]. Some inhabitants of Easter Island (Rapa Nui) carry HLA alleles characteristic of South America, most readily explained by gene flow after the colonization of the island around 1200 CE but before European contact in 1722 [5]. In Brazil, two nineteenth-century Botocudo skulls carrying the mtDNA Polynesian motif have been reported, and a Pre-Columbian date for entry of this motif into the Americas discussed, although a more recent date was considered more likely [6]. Thus South America was in two-way contact with other continental regions in prehistoric times, but there is currently no unequivocal evidence for outside gene flow into South America between the initial colonization by the ‘First American’ stream and European contact. The Y chromosome has a number of advantages for studies of human migrations. Haplotypes over most of its length are male-specific and evolve along stable lineages only by the accumulation of mutations, and the small male effective population size results in high levels of genetic drift of these haplotypes [7]. Consequently, major Y lineages (haplogroups) are often specific to particular geographical areas, and migrant haplogroups may stand out because they are atypical of the place where they are found (e.g. [8,9]). The most comprehensive study of indigenous South American Y chromosomes thus far surveyed 1011 individuals and found that while most of them belonged to haplogroup Q as expected, 14 individuals from two nearby populations in Ecuador carried haplogroup C3*(xC3a-f) chromosomes (henceforth C3*), with this haplogroup reaching 26% frequency in the Kichwa sample and 7.5% in the Waorani [10]. The estimated TMRCA for the combined Ecuadorian C3* chromosomes was 5.0–6.2 Kya. The finding of this haplogroup in Ecuador was surprising because C3* is otherwise unreported from the Americas (apart from one example in Alaska), but is widespread and common in East Asia. Three scenarios might explain the presence of C3* chromosomes at a mean frequency of 17% in these two Ecuadorian populations [10], Fig. 1. First, they might represent recent admixture with East Asians during the last few generations. This possibility was considered unlikely because the Waorani discouraged contact with outsiders using extreme ferocity until peaceful links were established in 1958, and known male ancestors (fathers, grandfathers) of C3* carriers were born before this date. Second, C3* might have been another founding lineage entering the Americas 15–20 Kya, and have drifted down to undetected levels in all populations examined except the Ecuadorians. This was also considered unlikely because the populations of North and Central America have in general experience less drift and retained more diversity than those in South America [2], and so it would be surprising to lose C3* from North/Central Americans but not South Americans. Third, C3* could have been introduced into Ecuador from East Asia at some intermediate date by a direct route that bypassed North America. In support of this third scenario, archaeologists have identified similarities in pottery between the middle Jōmon culture of Kyushu (Japan) and the Valdivia culture of coastal Ecuador dating to 5.3–6.4 Kya; notably, like the C3* chromosomes, such a ceramic complex in the Americas was unique to Ecuador and was not reported from North or Central America or elsewhere from South America [11]. We refer to these three scenarios as ‘recent admixture’, ‘founder plus drift’ and ‘ancient admixture’, respectively.  In this follow-up study, we set out to revisit the three hypotheses for the origin of the C3* Y chromosomes in Ecuador. One possibility would be to sequence the Ecuadorian C3* Y chromosomes, and compare them with existing or additional East Asian C3* chromosome sequences, to determine the divergence time. However, the limited quantity and quality of DNA available did not allow this. We therefore followed another possibility, using genome-wide autosomal SNP genotyping. By comparing the autosomal genotypes of the Kichwa and Waorani samples with other populations including Native South Americans and East Asians, we expect that admixture in the last few generations (‘recent admixture’) would be readily detectable, and admixture ∼6 Kya potentially detectable (‘ancient admixture’), from the presence of East Asian autosomal segments only found in the Ecuadorian samples compared with the other Native South Americans. This would allow discrimination between these hypotheses. With whole genome autosomal data, we could investigate the whole population samples of both females and males, including either carriers of C3* chromosomes or of other Y haplogroups, since any admixture would affect the whole population. |
|
|
|
Post by Admin on Apr 24, 2018 18:27:55 GMT
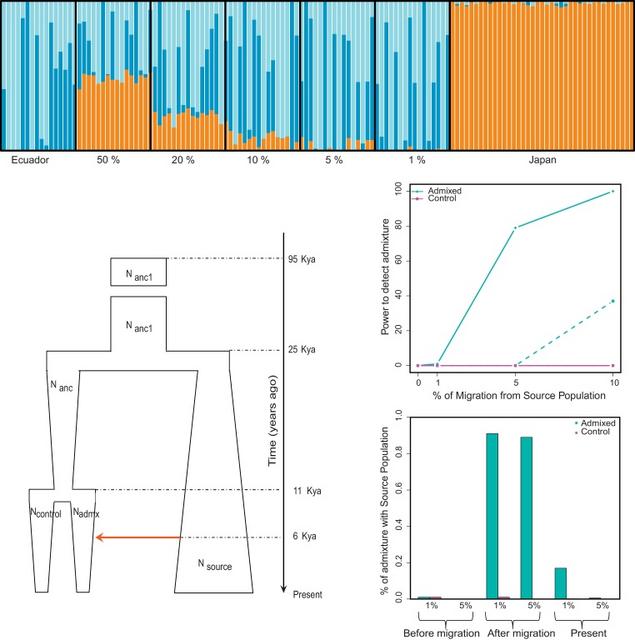 Fig. 2. Evaluation by simulation of the power to detect recent or 6 Kya Japanese admixture in the Ecuadorian population. (A) Simulation of recent admixture: ADMIXTURE analysis of artificially admixed Ecuadorian and Japanese genomes. For the optimal value of K (K = 3), the Japanese component (orange) is absent from the Ecuadorian population, but is present in some or all individuals from the admixed populations with admixture levels of 50%, 20%, 10%, 5% or 1%. (B)–(D) Simulation of admixture 6 Kya. (B) Demographic model used in the simulations. We considered one population of constant size for 70 Ky from 95 Kya to 25 Kya (with size Nanc1). At 25 Kya, this split into two. One daughter population (Nsource) expanded and represents the Source population. The other split into two 11 Kya, representing the Control population (Ncontrol) and the Admixed population (Nadmix). A single pulse of migration occurred 6 Kya (red arrow) from the Source to the Admixed population, with the level of admixture varied in different simulations. (C) Power to detect admixture that occurred 6 Kya in present-day samples. Migration 6 Kya was set at 0%, 1%, 5% or 10%. The cyan continuous line shows the power to detect a mean level ≥0.1% admixture in the present-day Admixed population, and the cyan dotted line the power to detect ≥0.5% admixture. The magenta lines show the corresponding detection in the Control population. (D) The fraction of simulated individuals from the scenario with 10% migration 6 Kya who have an admixture level greater than 1% or 5%, sampled before the migration, one generation after, or 6 Ky after in the present-day population: cyan, in the Admixed population; magenta, in the control population. For the recent admixture model, we found that we could detect ∼50% or ∼20% of Japanese ancestry in all the individuals in the 50% or 20% artificial admixed simulations, respectively. With lower proportions of admixture, there was more variation between individuals, but we identified 3–14% Japanese ancestry in all but one individual in the 10% artificial admixed simulation. We detected 1–9% of Japanese ancestry in about half of the individuals in the 5% artificial admixed simulations, and 1–2% in two individuals in the simulations of 1% artificial admixture (Fig. 2A). So we are well-powered for detecting recent admixture, and detect it in some individuals from a population sample of 16 even at 1% admixture. We then simulated a scenario where the admixture had occurred 6 Kya, using the demographic parameters estimated from the linkage disequilibrium pattern as described previously [20], shown in Fig. 2B and Supplementary Table 2. A single pulse of migration was set at 0%, 1%, 5% and 10%. Due to genetic drift in the relatively small population, after 6 Ky the population average level of admixture in the present-day population was much less than the starting amount. The power to detect ancient admixture at these levels therefore depends on the sensitivity to detect the reduced admixture in the present-day population. For example, if 0.1% mean population admixture can be detected in the present-day population, we have ∼80% power to detect 5% ancient admixture and ∼100% power to detect 10% ancient admixture. If, instead, we could only detect 0.5% mean admixture in the present-day population, we have ∼0 power to detect 5% ancient admixture and ∼35% power to detect 10% ancient admixture (Fig. 2C). With these population mean levels of admixture, the admixture in different individuals in the population can vary substantially. Immediately after a pulse of 10% migration, almost all individuals in the Admixed population have >5% and >1% admixture (Fig. 2D, middle section), as also seen in Fig. 2A. 6 Ky later, in the present-day population, about 1% of the Admixed individuals retain 5% admixture, and about 17% retain 1% admixture (Fig. 2d, right-hand section). In all simulations, negligible admixture is detected in the Control population. Overall, we have good power to detect 10% admixture that took place 6 Kya, and some power to detect 5% ancient admixture. |
|
|
|
Post by Admin on Apr 25, 2018 18:41:22 GMT
3.2. Population structure, admixture and migration patterns observed in Ecuadorians We genotyped the available Ecuadorian samples at ∼2.5 M sites. The quality of the DNA was low, so it was necessary to perform whole-genome amplification, and even after this step only about half (16/31) of the samples passed QC. For comparison, we analyzed 11 whole-genome amplified JPT samples in parallel. In order to assess the results, we first compared the genotypes with those of the HGDP populations, using either the worldwide set, or a subset focussed on the relevant populations. Worldwide (Supplementary Fig. 2) and focused PCA (Fig. 3A) both showed that the amplified JPT fell among the HGDP Japanese, indicating that the amplification procedure and different genotyping chip and centre had no effect detectable by this analysis. Similarly, the Ecuadorians grouped with other Native American populations. ADMIXTURE analyses supported these findings, although with the Ecuadorians tending to form their own cluster at the optimal value of K (Supplementary Fig. 3; Fig. 3B). Some Ecuadorian individuals showed evidence of ancestral components shared with other Native American populations, visible, for example, as the mid green and light green components in Fig. 3B. In addition, in the focussed analysis, two Ecuadorians showed around 5% of a pink ancestral component most prevalent in the Yakut. This component was also detectable at a low level in some Colombians and all of the Maya, as well as in the Russians, so may represent widespread ancient shared ancestry. None of the Ecuadorians showed any of the red component characteristic of the Japanese. This red component was, however, detectable in most of the Maya. 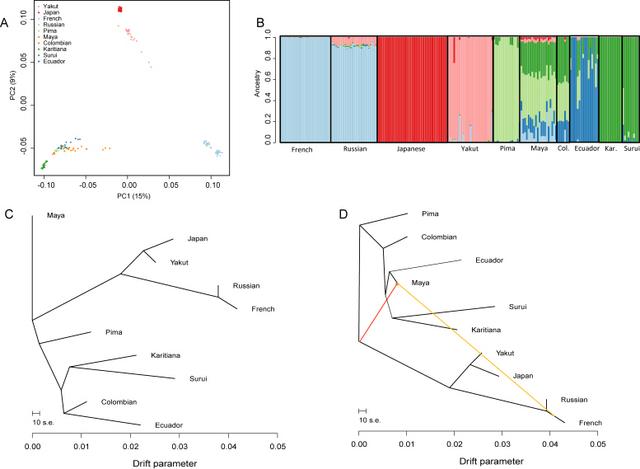 Fig. 3 Analysis of admixture and migration in the Ecuadorian, Japanese, and other relevant populations. (A) Principal Components Analysis of a focused dataset consisting of the Ecuadorians, two European populations, two East Asian populations, and all the Native American populations in the HGDP panel. (B) Admixture plot for the same populations with K = 6. The Japanese cluster was not detected in the Ecuadorians. (C, D) Migration graphs generated using TREEMIX. (C) TREEMIX with no migration. (D) TREEMIX with the maximum supported number of migration events (two) reveals two migration events into the Maya, but no evidence of migration into the Ecuadorians. We set out to test whether or not the haplogroup C3* Y chromosomes found at a mean frequency of 17% in two Ecuadorian populations [10] could have been introduced by migration from East Asia, where this haplogroup is common. We considered recent admixture in the last few generations and, based on an archaeological link between the middle Jōmon culture in Japan and the Valdivia culture in Ecuador [11], a specific example of ancient admixture between Japan and Ecuador 6 Kya (Fig. 1).  Simulations of recent admixture, and ancient admixture based on a demographic model of the relevant populations (Fig. 2B), revealed that we had good power to detect 1% recent admixture and 10% ancient admixture, with some power to detect 5% ancient admixture (Fig. 2). The lower power to detect ancient admixture was due to the extensive drift in the small Native American populations providing opportunities for the admixture signal to be lost by chance. No evidence for admixture was found in the autosomal SNP genotype data (Fig. 3, Table 1). Since the C3* Y chromosomes are present in the Ecuadorian populations at moderate frequency, the absence of evidence for >1% recent admixture is strong evidence against their recent introduction into Ecuador. It is more difficult to rule out ancient admixture. While no such admixture was detected, it remains possible that ancient admixture occurred at a low level (e.g. 1%), the introduced Y chromosomes then drifted up in frequency to their present level, and the introduced autosomal segments remained at, or drifted down to, undetectable levels. Nevertheless, the simplest interpretation of our results is that there was no ancient admixture, and the explanation for the presence of the C3* Y chromosomes in Ecuador must lie elsewhere. The remaining scenario is the ‘founder plus drift’ model (Fig. 1). With this model, the difficulty is to explain why the generally more genetically diverse North and Central American populations lack C3* Y chromosomes, while the less diverse South American populations retain them. Future simulations can be used to address this issue, and C3* Y chromosome with potential North/Central Native American affiliations should be evaluated carefully. Ancient DNA samples would be particularly relevant. In addition, as indicated in the Introduction, an attractive approach would be to sequence modern Ecuadorian and Asian C3* Y chromosomes and estimate the divergence time [23]: a time >15 Kya would support the founder plus drift model, while a time of 6 Kya or slightly higher would support the specific ancient admixture model considered here. Additional Ecuadorian DNA samples will be required for this. 5. Conclusions Three different hypotheses to explain the presence of C3* Y chromosomes in Ecuador but not elsewhere in the Americas were tested: recent admixture, ancient admixture ∼6 Kya, or entry as a founder haplogroup 15–20 Kya with subsequent loss by drift elsewhere. We can convincingly exclude the recent admixture model, and find no support for the ancient admixture scenario, although cannot completely exclude it. Overall, our analyses support the hypothesis that C3* Y chromosomes were present in the “First American” ancestral population, and have been lost by drift from most modern populations except the Ecuadorians. Forensic Sci Int Genet. 2015 Mar; 15: 115–120. doi: 10.1016/j.fsigen.2014.11.005 |
|
|
|
Post by Admin on Feb 4, 2021 21:17:21 GMT
 Alexis Sanchez went to the Mid-Columbia library in Kennewick expecting to pick up a book, but when she walked into the library, she stopped dead in her tracks. "I was like, 'why do they have a statue of my dad here?'" Sanchez said. "Like I literally thought it was my dad." She told NBC Right Now that she walked up closer to the bust and realized it was a display on Kennewick Man: the famous 9,000 year old skull found on the shore of the Columbia River in 1996. This inspired her to learn more about her heritage, so she took a 23andMe DNA test. She could only get the maternal haplogroup (the results from her mother's side), so a bit later she had her father, Javier Sanchez, submit his DNA for 23andMe as well. "Two years ago what they had for my dads haplogroup - it's like a paternal lineage - was that he was just related to Native Americans who traveled down to the Oregon coast... so that's why I was like, 'oh, that's why I feel so close; I love the Oregon coast,'" Sanchez said. For years, she has been teasing her father about looking like Kennewick Man. In May of 2013 she posted a side-by-side comparison photo of her father and Kennewick Man on Instagram.  Last month, she re-checked her 23andMe account. "I literally was just screaming on my couch!" she exclaimed. 23andMe had updated information linking her family to Kennwick Man. Essentially, her father shares paternal lineage with 'The Ancient One.' Javier said, "I did it and something came out with the Kennewick Man; it really surprised me. I'm from Mexico and I came here when I was 16 and a half years old." If you remember, scientists analyzed Kennewick Man's DNA and discovered in 2015 that Kennewick Man was Native American. Therefore, Javier and Kennewick Man belong to Haplogroup Q-M3. A haplogroup is a group of single DNA strands, that all share a common ancestor at some point - connecting a Kennewick man to Kennewick Man. "It's a surprise finding out that by finding out the DNA test I'm kind of a descendant of the Kennewick Man... it's amazing, that surprised me a lot," Javier Sanchez said. |
|
|
|
Post by Admin on Feb 5, 2021 4:10:47 GMT
The ancestry and affiliations of Kennewick Man Morten Rasmussen, Martin Sikora, Anders Albrechtsen, Thorfinn Sand Korneliussen, J. Víctor Moreno-Mayar, G. David Poznik, Christoph P. E. Zollikofer, Marcia S. Ponce de León, Morten E. Allentoft, Ida Moltke, Hákon Jónsson, Cristina Valdiosera, Ripan S. Malhi, Ludovic Orlando, Carlos D. Bustamante, Thomas W. Stafford Jr, David J. Meltzer, Rasmus Nielsen & Eske Willerslev Nature volume 523, 455–458(2015) Abstract Kennewick Man, referred to as the Ancient One by Native Americans, is a male human skeleton discovered in Washington state (USA) in 1996 and initially radiocarbon dated to 8,340–9,200 calibrated years before present (BP)1. His population affinities have been the subject of scientific debate and legal controversy. Based on an initial study of cranial morphology it was asserted that Kennewick Man was neither Native American nor closely related to the claimant Plateau tribes of the Pacific Northwest, who claimed ancestral relationship and requested repatriation under the Native American Graves Protection and Repatriation Act (NAGPRA). The morphological analysis was important to judicial decisions that Kennewick Man was not Native American and that therefore NAGPRA did not apply. Instead of repatriation, additional studies of the remains were permitted2. Subsequent craniometric analysis affirmed Kennewick Man to be more closely related to circumpacific groups such as the Ainu and Polynesians than he is to modern Native Americans2. In order to resolve Kennewick Man’s ancestry and affiliations, we have sequenced his genome to ∼1× coverage and compared it to worldwide genomic data including for the Ainu and Polynesians. We find that Kennewick Man is closer to modern Native Americans than to any other population worldwide. Among the Native American groups for whom genome-wide data are available for comparison, several seem to be descended from a population closely related to that of Kennewick Man, including the Confederated Tribes of the Colville Reservation (Colville), one of the five tribes claiming Kennewick Man. We revisit the cranial analyses and find that, as opposed to genome-wide comparisons, it is not possible on that basis to affiliate Kennewick Man to specific contemporary groups. We therefore conclude based on genetic comparisons that Kennewick Man shows continuity with Native North Americans over at least the last eight millennia. Main The skeleton of Kennewick Man was inadvertently discovered in July of 1996 in shallow water along the Columbia River shoreline outside Kennewick, Washington state, USA. On several visits to the locality over the following month, some 300 bone elements and fragments were collected, ultimately comprising ∼90% of an adult male human skeleton3. The initial assessment of this individual was that he was a historic-period Euro-American, based largely on his apparently “Caucasoid-like”3 cranium, along with a few artefacts found nearby (later proved not to be associated with the skeletal remains). However, radiocarbon dating subsequently put the age of the skeleton in the Early Holocene1. The claim that Kennewick Man was anatomically distinct from modern Native Americans in general, and in particular from those tribes inhabiting northwest North America4, sparked a legal battle over the disposition of the skeletal remains. Five tribes who inhabit that region requested the remains be returned to them for reburial under the Native American Graves Protection and Repatriation Act (NAGPRA). The US Army Corps of Engineers, which manages the land where Kennewick Man was found, announced their intent to do so. That in turn prompted a lawsuit to block the repatriation2,5, and generated considerable scientific controversy as to Kennewick Man’s ancestry and affinities (for example, refs 3, 6, 7, 8, 9). The lawsuit ultimately (in 2004) resulted in a judicial ruling in favour of a detailed study of the skeletal remains, the results of which were recently published2. These studies provide important details on, for example, Kennewick Man’s life history, refine his antiquity to 8,358 ± 21 14C years BP or to within a two sigma range of 8,400–8,690 calibrated years BP (based on 90% marine diet, and 750 year marine reservoir correction), and demonstrate that the body had been intentionally buried and had eroded out shortly before discovery2. They also include anatomical and morphometric analyses, which confirm earlier studies that Kennewick Man resembles circumpacific populations, particularly the Ainu and Polynesians2,10; that he has certain “European-like morphological” traits2; and that he is anatomically distinct from modern Native Americans2. These results are interpreted as indicating that Kennewick Man was a descendant of a population that migrated earlier than, and independently of, the population(s) that gave rise to modern Native Americans2. However, those recent studies did not include DNA analysis. Herein we present the genome sequence of Kennewick Man in order to resolve his ancestry and affinities with modern Native Americans. There were several prior efforts to recover genetic material from Kennewick Man11, but none were successful. We obtained ∼1 × coverage of the genome, from 200 mg of metacarpal bone specimen (Supplementary Information 1) using previously published methods12,13. The endogenous DNA content was between 0.4% and 1.4% for double-stranded and single-stranded libraries, respectively (Supplementary Information 2). Average fragment length was 53.6 base pairs (bp) and the sample exhibited damage patterns typical of ancient DNA, with excessive deamination of cytosine towards the ends of the fragments (Supplementary Information 2). Similarly, patterns of DNA decay agree with published expectations14, and display an estimated molecular half-life corresponding to 3,670 years for 100-bp molecules (Supplementary Information 3). The mitochondrial genome was sequenced to ∼71× coverage and is placed at the root of haplogroup X2a (Extended Data Fig. 1 and Supplementary Information 2), and the Y-chromosome haplogroup is Q-M3 (Extended Data Fig. 2 and Supplementary Information 5); both uniparental lineages are found almost exclusively among contemporary Native Americans15,16. We used the X chromosome to conservatively estimate contamination to be 2.5%, which is within the normal range obtained observed in genomic data from ancient human remains17, and we further show this contamination to be of European origin (Supplementary Information 4). We compiled an autosomal reference data set consisting of published SNP array data18,19,20,21,22,23 as well as new data generated from one of the claimant tribes, the Colville (Supplementary Information 10). Due to high levels of recent admixture in many Native American populations, we masked European ancestry from the Native Americans (Supplementary Information 6). No masking was done on the Kennewick Man. When we compare Kennewick Man with the worldwide panel of populations, a clear genetic similarity to Native Americans is observed both in principal components analysis (PCA) and using f3-outgroup statistics (Fig. 1a, b). In particular, we can reject the hypothesis that Kennewick Man is more closely related to Ainu or Polynesians than he is to Native Americans, as seen in a D-statistic-based test where no trees of the type ((CHB,Ainu/Polynesian),(X,Karitiana)) with X being Kennewick Man, the Clovis age Anzick-1 child (ref. 12) or a modern Native American genome are rejected (Extended Data Fig. 3). Model-based clustering using ADMIXTURE24 shows that Kennewick Man has ancestry proportions most similar to those of other Northern Native Americans (Fig. 1c and Supplementary Information 7), especially the Colville, Ojibwa, and Algonquin. Considering the Americas only, f3-outgroup and D-statistic based analyses show that Kennewick Man, like the Anzick-1 child, shares a high degree of ancestry with Native Americans from Central and South America, and that Kennewick Man also groups with geographically close tribes including the Colville (Fig. 2a, b and Extended Data Fig. 4). Despite this similarity, Anzick-1 and Kennewick Man have dissimilar genetic affinities to contemporary Native Americans. In particular, we find that Anzick-1 is more closely related to Central/Southern Native Americans than is Kennewick Man (Extended Data Fig. 5). The pattern observed in Kennewick Man is mirrored in the Colville, who also show a high affinity with Southern populations (Fig. 2c), but are most closely related to a neighbouring population in the data set (Stswecem’c; Extended Data Fig. 4c). This is in contrast to other populations such as the Chipewyan, who are more closely related to Northern Native Americans rather than to Central/Southern Native Americans in all comparisons (Fig. 2d and Extended Data Fig. 4d). 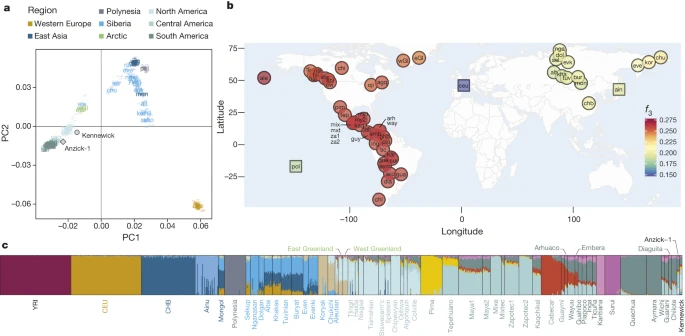 Figure 1: Genetic affinities between Kennewick Man and a panel of world-wide populations. |
|

