|
|
Post by Admin on Jun 21, 2022 17:50:10 GMT
EBA—Únětice culture
The transition to the EBA in Bohemia is associated with a positive shift in the coordinates of PC2, relative to preceding late BBs (Fig. 4B, fig. S7, and table S31). Admixture f3 statistics are most negative when EHG (Eastern HG) or WSHG (West Siberian HG) are used as a second source in addition to the geographically and temporally proximal Bohemia_BB_Late (table S32), suggesting a northeastern contribution to Bohemia_Únětice_preClassical. To find a suitable proxy for a potential additional source population, we modeled Bohemia_Únětice_preClassical as a two-way mixture of local Bohemia_BB_Late and various sources more positive on PC2 (table S33). We reject mixture models involving Bohemia_BB_Late and Yamnaya (Samara, P = 5.3 × 10−10; Kalmykia, P = 5.8 × 10−10; Ukraine, P = 7.3 × 10−12; and Caucasus, P = 3.2 × 10−15) or Bohemia_BB_Late and CW (early, P = 1.1 × 10−4; late, P = 5.4 × 10−6). We fail to reject a two-way mixture model of 63.5% Bohemia_BB_Early and 36.5% Bohemia_BB_Late (P = 0.29), suggesting a large (63.5%) contribution from an early BB lineage, which was largely unsampled during the late BB phase (2400 to 2200 BCE), but represents a potential new lineage at the dawn of the Bronze Age. The Y-chromosomal data suggest an even larger turnover. A decrease of Y-lineage R1b-P312 from 100% (in late BB) to 20% (in preclassical Únětice) implies a minimum 80% influx of new Y-lineages at the onset of the EBA.
However, aware of the limited resolution of Bohemia_BB_Early (small sample size, low resolution, and large SEs), we explored alternative models for preclassical Únětice individuals. All model fits improve when Latvia_BA is included in the sources, resulting in two additional supported models (table S33). A three-way mixture of Bohemia_BB_Late, Bohemia_CW_Early, and Latvia_BA (P value of 0.086) not only supports a more conservative estimate of 47.7% population replacement but also accounts for the Y-chromosomal diversity found in preclassical Únětice, with R1b-P312 from Bohemia_BB_Late, R1b-U106 and I2 from Bohemia_CW_Early, and R1a-Z645 from Latvia_BA (Fig. 4A).
Although the geographic origin of this new ancestry cannot be precisely located, three observations offer clues. First, the Latvia_BA ancestry that improves all model fits (table S33) suggests an ultimate northeastern origin. Second, Y-haplogroup R1a-Z645 appears in Bohemia (and wider central Europe) for the first time at the beginning of the EBA, a lineage previously fixed in Baltic and common in Scandinavian CW males (23, 24), supporting a north/northeastern genetic contribution. Third, an Únětice genetic outlier (VLI051, male, Y-haplogroup R1a-Z645; table S34) resembles individuals from Bronze Age Latvia (Fig. 2D) (68), providing direct evidence for migrants from the northeast.
We also detect a genetic shift in the transition from preclassical to classical Únětice, reflected in the decrease in PC2 coordinates for Únětice individuals dated after ~2000 BCE (Fig. 4B and fig. S7) and confirmed using qpWave (table S35) and f4 statistics (table S36). Bohemia_Únětice_Classical can be modeled as a mixture of Bohemia_Únětice_preClassical and a local Eneolithic source (table S37). In contrast to the genetic shift between late BB and preclassical Únětice, the Y-lineage diversity remains similar throughout both Únětice phases, suggesting assimilation and subtler social changes.
|
|
|
|
Post by Admin on Jun 22, 2022 15:49:50 GMT
DISCUSSION The high-resolution genetic time transect in Bohemia, allowing early and late phases of cultural groups to be divided and studied separately (e.g., CW, BB, and Únětice), elucidates several major processes before and after the arrival of steppe ancestry (Fig. 6). Our dense sampling allows detection of novel, important, and perhaps “unexpected” changes within cultural groups (e.g., CW and BB), if they are seen through a strict cultural-historical lens. Previous studies have largely been interpreted as revealing major migrations at the beginning and end of the Neolithic (i.e., periods where the incoming groups were genetically very distinct); however, our results reveal additional large genetic turnovers. By sampling consecutive and partially contemporaneous cultural groups, we show that the spread of Funnelbeaker and GAC (69, 70), as well as the origin of Únětice, involved large genetic shifts over short time periods, likely explained by migrations.  Fig. 6. Schematic summary of the major processes that shaped the genetic and cultural diversity of Bohemia (red outline) over time. Arrows on maps indicate a general direction of influences rather than discrete routes of migration. We show that early CW were genetically exceptionally diverse, some resembling GAC and Yamnaya, with a few also falling outside of previously sampled central European Neolithic genetic diversity. Such a notably diverse signal is likely the result of the agglomeration of people from diverse cultural and linguistic backgrounds into an archaeologically similar but polyethnic or plural society. Important factors in ethnic identity include ancestry, history, ideology, and language (71, 72). The level of genetic differentiation (i.e., time since common ancestor) between early CW individuals with high and no steppe ancestry implies long biological isolation and hence different histories. The finding of GAC-like and Yamnaya-like genetic profiles in early CW suggests integration of people who came from ideologically diverse societies (i.e., neither GAC nor Yamnaya practiced strong gender differentiation in mortuary practices, unlike CW). It is likely that GAC and CW/Yamnaya individuals spoke different languages (3, 4, 43), meaning that early CW society in Bohemia encompassed people who had demonstrably different histories, likely originating from ideologically diverse cultures, who spoke different mother tongues. The assimilation process of individuals without steppe ancestry into early CW society was female-biased (43). However, finding females also among individuals with the highest amounts of steppe ancestry (3 of 5; Fig. 2B) suggests that they were also well represented among migrating CW individuals [in contrast to (43)] or perhaps assimilated from nearby Yamnaya groups (e.g., Hungary). Finding individuals without steppe ancestry in early CW contexts (n = 4) is more common than individuals with steppe ancestry in pre-CW contexts (e.g., GAC, n = 0). This pattern of asymmetric gene flow between the contemporaneous GAC and CW may reflect newcomers (CW groups) having more benefit from incorporating people with important local knowledge (i.e., from pre-CW cultural contexts) into their communities. The archaeological record shows continuity of such knowledge (e.g., pottery production and lithic raw materials) in several regions (22, 67, 73). Vliněves is crucial for elucidating interactions between individuals with high and no steppe ancestry. This site yields the earliest dated CW (VLI076, 3018 to 2901 BCE) who is also genetically most differentiated from pre-CW individuals, while 20% (3 of 15) of the sampled early CW from Vliněves had no steppe ancestry. Intriguingly, we observe no archaeological differences between CW graves of individuals with and without steppe ancestry from two sites (Vliněves and Stadice; see the Supplementary Materials), suggesting full integration of genetically, and likely ethnically, diverse individuals within the same archaeological culture. Finding Latvia_MN-like ancestry in early CW, in conjunction with the absence of Y-chromosomal sharing between early CW and Yamnaya males, suggests a limited or indirect role of known Yamnaya in the origin and spread of CW to central Europe. Our results allude to either a northeast European Eneolithic forest steppe contribution to early CW or a hitherto unsampled steppe population who carried excess Latvia_MN-like ancestry, a scenario that is less likely given the high degree of genetic homogeneity among 3000-BCE steppe groups [e.g., Yamnaya and Afanasievo separated by ~2500 km but genetically almost indistinguishable (4, 61)]. As much of 4000- to 2500-BCE (north)eastern Europe remains unsampled, inferring the precise geographic origin of early CW individuals remains elusive.
Since social kinship systems influence patterns of genetic diversity (13, 42, 48, 74), it is likely that several different kin systems existed in third millennium BCE central Europe. The highly diverse genetic profiles (both nuclear and Y-chromosomal) of early CW suggest a different social organization to late CW and BB, whose Y chromosome pattern is indicative of strict patrilineality. This suggests that different cultural groups, in addition to using various forms of material culture and mortuary practices, likely also conformed to different ideologies as expressed in their mating pattern and/or social organization. This is supported by the finding of completely nonoverlapping Y chromosome variation between the partially contemporaneous late CW and BB, indicating a large degree of paternal mating isolation between these two groups, even when found at the same site (e.g., Vliněves).
The onset of the preclassical Únětice was accompanied by a ≥40% nuclear and ≥80% Y-chromosomal contribution ultimately originating from the northeast and breaking down the gender-differentiated mortuary practices and strict patrilineality of late CW and BB. This was neither evident in the burial customs nor in the material culture but could represent the underlying connection to the Baltics, the ultimate source of EBA amber in Bohemia associated with the later emerging Amber Road (75–77). Therefore, our results suggest two main periods (early CW and early Únětice) of genetic influence from the northeast, much of which remains unsampled in the European archaeogenetic record (e.g., Belarus).
Our results reveal a complex and highly dynamic history of Neolithic to EBA central Europe, during which migration and the movement of people facilitated abrupt genetic and social changes. Large-scale demic expansions occurred multiple times before and after the appearance of steppe ancestry in Europe. Early CW society was diverse and emerged amid a strong cultural and genetic transition, involving males and females of diverse origins and likely ethnicities. Genetic shifts occurred within CW, BB, and EBA societies despite continuity in material culture. Cultural affiliations played a major role in third millennium BCE social behaviors, which ultimately changed with the influx of new people over time. Although the impact of social processes is observable in patterns of genetic diversity, further interdisciplinary research is required to characterize the drivers of these changes, both at a micro- and macro-regional level.
|
|
|
|
Post by Admin on Jun 23, 2022 16:46:34 GMT
MATERIALS AND METHODS
Processing sites for the newly reported individuals
Most (186 of 206, 90.2%) of the newly reported individuals were entirely processed at the Max Planck Institute for the Science of Human History in Jena, Germany, and the full details of sampling and ancient DNA wet laboratory work and bioinformatic processing are summarized in what follows. The individuals from the site Makotřasy were initially sampled and processed into powder at the University of Vienna, followed by subsequent laboratory work and bioinformatic and ancient DNA analysis at Harvard Medical School following previously described protocols (61).
Sampling
In total, 389 pars petrosa, teeth, and bones from 261 individuals were processed as part of this study. Upon introduction into the clean room facilities at the Max Planck Institute for the Science of Human History in Jena, Germany, all samples were wiped with 5% bleach and ultraviolet (UV) irradiated for 20 min on each side. Teeth were sampled by removing the crown followed by drilling into the pulp chamber to create bone powder. Pars petrosa were sampled by drilling into their dense region (78) to create bone powder. Between 50 and 100 mg of resulting bone powder from each sample were collected in different 2-ml Biopure tubes (one tube per sample) and used in subsequent DNA extraction.
DNA extraction
One milliliter of extraction buffer [containing 0.9 ml of 0.5 M EDTA, 0.025 ml of proteinase K (0.25 mg/ml), and 0.075 ml of UV high-performance liquid chromatography (HPLC) water] was added to Biopure tubes containing bone powder. Biopure tubes were then sealed with Parafilm and incubated overnight on a rotating wheel at 37°C. After incubation, Biopure tubes were spun for 2 min at 18,500 relative centrifugal force (rcf), separating the soluble from insoluble parts of the resulting solution. The soluble part was transferred to a 50-ml falcon tube containing 10 ml of binding buffer and 400 μl of sodium acetate (3 M, pH 5.2). Resulting mixture was transferred to a high pure extender assembly (HPEA) falcon tube, which was centrifuged at 1500 rpm in a 50-ml Thermo Fisher Scientific TX-400 Swinging Bucket Rotor for 8 min. The column from each HPEA tube was removed and inserted into a fresh collection tube and centrifuged at 18,500 rcf for 2 min. A total of 450 μl of wash buffer from the high pure viral nucleic acid kit (HPVNAK) was added to each column, which was then centrifuged at 8000 rcf for 1 min. Columns were then removed and placed into new collection tubes. Another round of washing was performed whereby 450 μl of wash buffer from the HPVNAK was added to each column and centrifuged at 8000 rcf for 1 min. Columns containing washed DNA were then transferred to 1.5-ml siliconized tubes. Fifty microliters of TET (tris-EDTA + Tween 20) buffer were added to the center of columns, and the columns were then incubated at room temperature for 3 min and centrifuged at 18,500 rcf for 1 min. Another 50 μl of TET buffer was added to the center of columns, after which they were centrifuged once more at 18,500 rcf for 1 min. The resulting 100 μl of DNA extracts was stored at −20°C until further processing.
DNA libraries and in-solution capture
Twenty-five microliters of DNA extract was used for the construction of (in most cases) double-stranded uracil DNA glycosylase (UDG) half-treated DNA libraries. UDG repair was performed by adding DNA extract to a 25-μl mastermix containing 6 μl of 10× Buffer Tango, 6 μl of 10 mM adenosine 5′-triphosphate, 0.5 μl of bovine serum albumin (BSA; 20 mg/ml), 0.2 μl of 25 mM each deoxynucleoside triphosphate (dNTP), 3.6 μl of 1-U USER enzyme, and 8.7 μl of UV HPLC water. Resulting mixture was incubated at 37°C for 30 min followed by 12°C for 1 min. Inhibition of UDG treatment was achieved through the addition of 3.6 μl of 2-U uracil glycosylase inhibitor (UGI) to each tube followed by incubation at 37°C for 30 min and again at 12°C for 1 min. Blunt-end repair was performed by adding 3 μl of 10-U T4 polynucleotide kinase and 1.65 μl of 3-U T4 DNA polymerase and incubating the resulting mixture at 25°C for 20 min and then at 12°C for 10 min. Blunt-end repaired mixture was purified using a MinElute kit and eluted in 20 μl of elution buffer (EB) containing 0.05% Tween. Illumina adapters were ligated onto DNA molecules through the mixture of 18 μl of eluate from the previous step with 20 μl of 2× Quick Ligase Buffer, 1 μl of 10 μM Adapter Mix, and 1 μl of 5-U Quick Ligase. Resulting mixture was incubated at 22°C for 20 min followed by purification with a MinElute kit and elution in 22 μl of EB containing 0.05% Tween. Adapter fill in reaction was performed by adding 20 μl of eluate from the previous step to 4 μl of 10× isothermal buffer, 0.2 μl of 25 mM each dNTP, 2 μl of 8-U Bst 2.0 polymerase, and 13.8 μl of UV HPLC water, followed by incubation at 37°C for 30 min and 80°C for 10 min. Resulting libraries were stored at −20°C until further processing. Unique library-specific indexes were added to the 5′ and 3′ ends of molecules in each library through an indexing polymerase chain reaction (PCR). Each library was split into four separate indexing PCR reactions, which were carried out using 10 μl of 10× Pfu Turbo buffer, 1.5 μl of BSA (20 mg/ml), 1 μl of 25 mM each dNTP, 1 μl of 2.5-U Pfu Turbo polymerase, 73.5 μl of UV HPLC water, 2 μl of 10 μM P5 index, 2 μl of 10 μM P7 index, and 9 μl of DNA library. Amplification was achieved through an initial denaturation at 95°C for 2 min, followed by 10 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 1 min, followed by 72°C for 10 min. Resulting indexed libraries of the same sample were pooled and purified using a MinElute purification kit. Purified libraries were quantified using quantitative PCR and amplified to 1013 copies. Amplified libraries were shallow shotgun sequenced (~5 million reads) on an Illumina HiSeq or NextSeq platform to estimate the general human DNA content, presence of ancient DNA damage, and mitochondrial:nuclear coverage ratio. Libraries with >0.1% endogenous human DNA and >5% C-to-T misincorporations at the 5′ end were chosen for 1240k capture (8) and mitochondrial DNA (mtDNA) capture. In cases where more than one library from the same individual satisfied the criteria for capture, the better quality (higher endogenous DNA content) library was used for 1240k and mtDNA capture.
Sequencing
Postcapture libraries were single-end (75 cycles) or paired-end sequenced (2 × 50 or 2 × 75) on HiSeq or NextSeq Illumina platforms to a depth of 20 to 50 million reads per library. Resulting sequence data were processed through EAGER (v1.92.38) (79). Illumina adapters were removed using AdapterRemoval (v2.2.0) (80), and in case of paired-end sequencing, corresponding reads from the same template molecule with a minimum of 11 base pairs (bp) of overlap were merged. Fastq files of merged and unmerged reads were concatenated, and reads shorter than 30 bp were discarded. Processed reads were mapped to the human reference genome (hg19) using BWA-aln and BWA-samse (v0.7.12) (81) applying maxdiff (-n) 0.01 and seeding turned off (-l 10000). Resulting bam files were sorted, and duplicate reads were removed using DeDup (v0.12.1) (https://github.com/apeltzer/DeDup). Damageprofiler (v0.3.10) (https://github.com/Integrative-Transcriptomics/DamageProfiler) was used to calculate rates of misincorporation in read termini of DNA fragments in our captured libraries. BAM files had the last three bases from both 5′ and 3′ ends of reads and corresponding base quality scores masked for downstream analyses (82).
Sex determination and authentication
The genetic sex of each sample (bam file) was determined by calculating the normalized mean coverage on the X (mean X coverage/mean autosome coverage) and Y (mean Y coverage/mean autosomal coverage) chromosomes (83). Samples with normalized mean Y coverage values greater than 0.2 were assigned male. Contamination was estimated in males by calculating the rate of heterozygosity on their X chromosome (84). In addition, we used Schmutzi to estimate the mitochondrial contamination in all libraries (85). Schmutzi was run on BAM files resulting from mapping 1240K capture sequencing data to the human mitochondrial reference genome. In cases where 1240k data were not enough to give an mtDNA contamination, we ran Schmutzi on the mtDNA capture data mapped to the human mitochondrial reference genome.
|
|
|
|
Post by Admin on Jun 23, 2022 19:43:58 GMT
Genotyping We used SAMtools (v1.3) (86) mpileup and pileupCaller from the sequenceTools (v1.4.0.2) package (https://github.com/stschiff/sequenceTools) to call pseudo diploid genotypes by sampling a random high-quality allele (base quality, ≥30; mapping quality, ≥30) from each of the 1240k sites (8). Newly generated genotype data for this study were then merged to a compiled dataset of previously published ancient and modern worldwide populations (https://reich.hms.harvard.edu/allen-ancient-dna-resource-aadr-downloadable-genotypes-present-day-and-ancient-dna-data) (v42.2) using mergeit (v2450) from the EIGENSOFT package (https://github.com/DReichLab/EIG). Mitochondrial and Y chromosome haplogroups Mitochondrial haplogroups were called by mapping 1240k or mtDNA capture data to the human mitochondrial reference genome followed by creating pileups at each position (map quality and base quality filter of 30) and calling the most frequent base at each position. Resulting genotype information was converted to fasta files, and haplogroups were called using haplofind (87). Y chromosome haplogroups were called by mapping 1240k capture data to the whole human reference genome (hg19) followed by visual inspection of ancestral/derived alleles (after map quality and base quality filter of 30) at ISOGG (v15.58 April 2020) sites. Principal components analysis PCA was conducted using smartpca (v1600) from the Eigensoft package (https://github.com/DReichLab/EIG). Principal components were calculated on the genotype data of modern West Eurasian individuals (table S7) (88–90). Ancient individuals were projected (lsqproject: YES) onto the axes calculated from modern individuals. “shrinkmode: YES” was used to account for artificial stretching of principal component axes between projected (ancient) individuals and modern individuals. Fst values were also calculated in smartpca using “fsthiprecision: YES” and “inbreed: YES” parameters. Ancestry decomposition and admixture modeling F statistics, qpWave, and qpAdm runs were conducted in admixr v0.7.1 (91), a wrapper program around ADMIXTOOLS (88). Selection of outgroups for each analysis is indicated in the corresponding Supplementary Table. Linear modeling of pre-CW HG ancestry (Fig. 3A) was performed using segmented linear regression as implemented in R using the segmented function for v.1.2-0 of the segmented library (92). To select the optimal number of breakpoints, we compared Akaike’s information criterion (AIC) for models with between zero and four breakpoints. The AIC is a score that considers how well a model fits the data while simultaneously penalizing models with additional parameters. In this way, model fit must be significantly improved for a more complicated model with additional parameters to be accepted over a simpler, nested model. A linear regression model with one breakpoint was found to have the minimum AIC and hence was selected (93). DATES v753 (61) was used to estimate length distributions of ancestry tracts and infer admixture dates between Anatolia_Neolithic and WHG (here, Loschbour+Körös_HG+Germany_BDB). Parameters binsize 0.001, maxdis 1, seed 77, jackknife YES, qbin 10, runfit YES, afffit YES, lovalfit 0.45, minparentcount 1, and checkmap YES were used. Y haplogroup frequency simulations To investigate the process of Y-haplogroup inheritance in early and late CW groups, we simulated 106 realizations, assuming a generational time of 25 years, and analyzed the results using approximate Bayesian computation (ABC). For the ith realization, we assumed a constant population size of Ni ~ U(102,104), with a starting a proportion of R1a-M417(xZ645) of pi ~ TN(0, 1)(0.27,0.134) from a truncated normal distribution based on the observed proportion of R1a-M417(xZ645) of 3 of 11 (0.27). For each simulation, we also included a selection coefficient denoted si ~ U(−1,1). Under random mating, for generation j + 1, let the number of a male offspring carrying R1a-M417(xZ645) be Xij+1 ~ B(Ni, wj), where wj = Xj−1/Nj−1. However, if one includes a selection coefficient, then wj = min(1,(1 + si)Xj−1/Nj−1). Hence, one may interpret sj as the average increase in the proportion of male offspring that R1a-M417(xZ645) individuals were having over this period. We then compared our observed number of per-generation R1a-M417(xZ645) to our simulated realizations using the rejection method and keeping the top closest 0.05% realizations (selected via cross-validation) to form samples from the joint posterior distributions for our simulation parameters. All ABC and cross-validation analyses were performed in R using the abc package (94). Supplementary Materials This PDF file includes: Supplementary Text Table S38 Figs. S1 to S9 Legends for tables S1 to S37 References DOWNLOAD 7.97 MB www.science.org/doi/suppl/10.1126/sciadv.abi6941/suppl_file/sciadv.abi6941_SM.pdf |
|
|
|
Post by Admin on Jun 24, 2022 19:29:38 GMT
The Únětice culture (ca. 2300-1700 BC) has been cited as a pan-European cultural phenomenon[Kristiansen and Larsson 2005], whose influence covered large areas due to intensive exchange[Pokutta 2013], with Únětice pottery and bronze artefacts found from Ireland to Scandinavia, the Italian Peninsula, and the Balkans. As such, it is candidate for a late community connecting a continuum of already scattered North-West Indo-European languages ancestral to Italic, Celtic, Germanic, and to Balto-Slavic, where words were frequently exchanged, sharing a common lexicon and certain regional isoglosses[Gamkrelidze and Ivanov 1995]. At the same time, strong phonetic differences found early in North-West Indo-European dialects, especially in the compounds with sonorants[Adrados, Bernabé, and Mendoza 2010][Clackson 2007], signal a period of already differentiated but inter-connected communities. 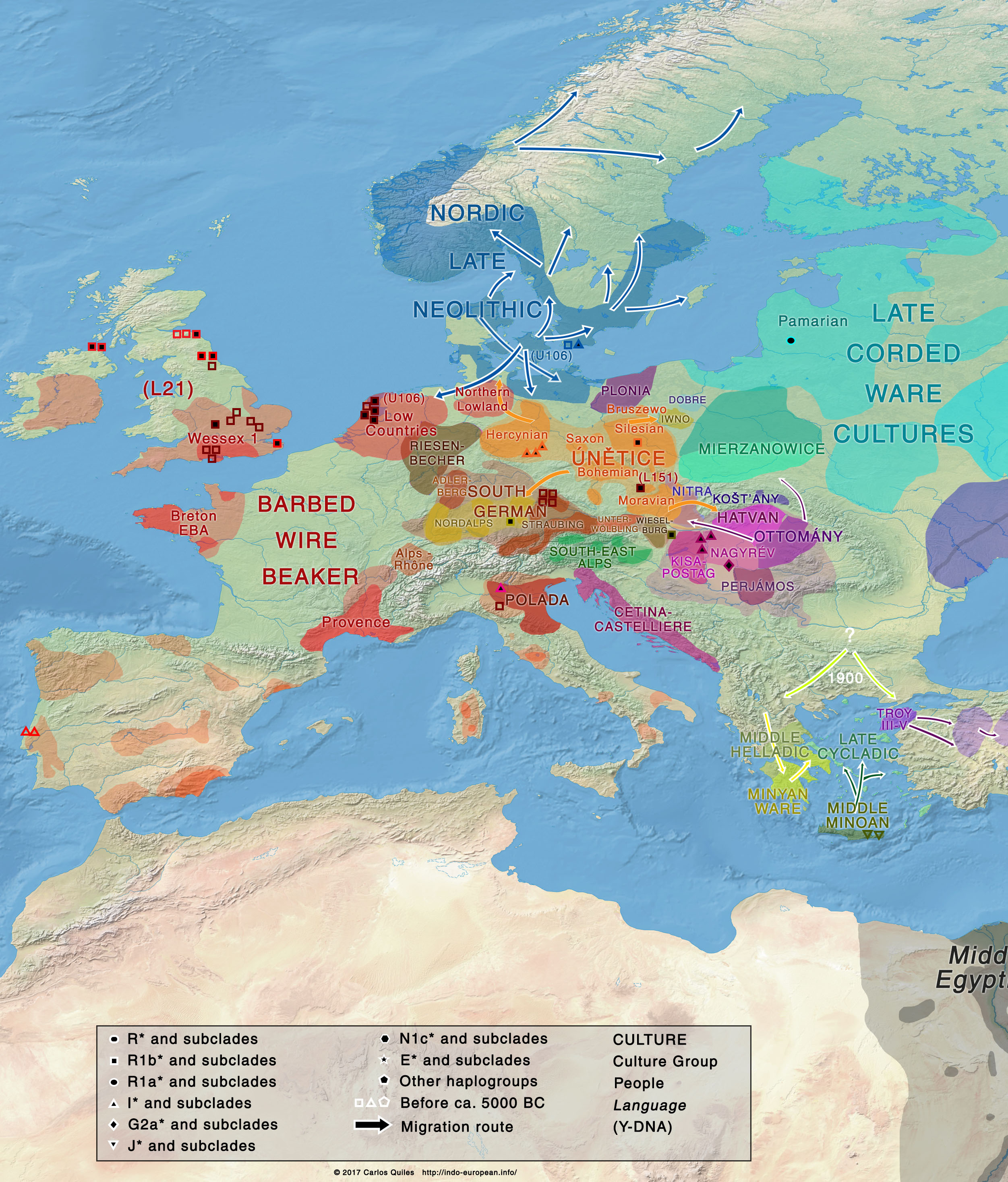 Thought to have evolved from Bell Beaker cultures, the scarce ancient Y-DNA available comes from four samples: one individual from Corded Ware/Proto-Únětice culture of R1b1a-L755 lineage at Łęki Małe ca. 2170 BC[Mathieson et al. 2017], and three from Únětice proper, of typical European hunter gatherer I2-M38 subclades – one ca. 2050 BC from Eulau, and two from Esperstedt dated ca. 2055 BC and 2035 BC[Mathieson et al. 2015]. On the periphery of the Únětice culture territory, haplogroup R1b1a1a2-M269 is found in Gata/Wieselburg (ca. 1765 BC), and haplogroup R1b1a1a2a1a-L151 in Untermeitingen ca. 1605 BC[Allentoft et al. 2015]. Ancient DNA samples suggest at least a partial resurge of hunter-gatherer ancestry in Únětice, although only a slightly lesser genetic affinity to Yamna than in Bell Beaker groups[Haak et al. 2015]. Úněticean genetic melting pot strengthens its origin as the vector of cultural diffusion of North-West Indo-European languages, essentially connecting Barbed Wire Beaker cultures from the Low Countries and the Northern Lowlands (and late Nordic Neolithic) – probably speaking languages ancestral to Germanic – with peoples of Southern German cultures, as predecessors of core regions of the Tumulus culture – possibly speaking West Indo-European, i.e. Pre-Italo-Celtic[Mallory 2013]. This suggests that Únětice connected these with eastern cultures like south-eastern European cultures – heirs of Bell Beaker and Carpathian groups – and the eastern Mierzanowice/Nitra culture – heir of Bell Beaker and Corded Ware groups. Therefore, the language ancestral to Balto-Slavic was probably spoken either by the Únětice population, or by eastern cultures that were connected to western Indo-European languages through Únětice. Bell Beakers and early Únětice represented the first prospectors and metallurgists, travelling and sharing their skills, with Adlerberg and Straubing groups of the Southern German cultures being small local centres[Kristiansen 1987]. |
|

