|
|
Post by Admin on Aug 11, 2017 19:46:58 GMT
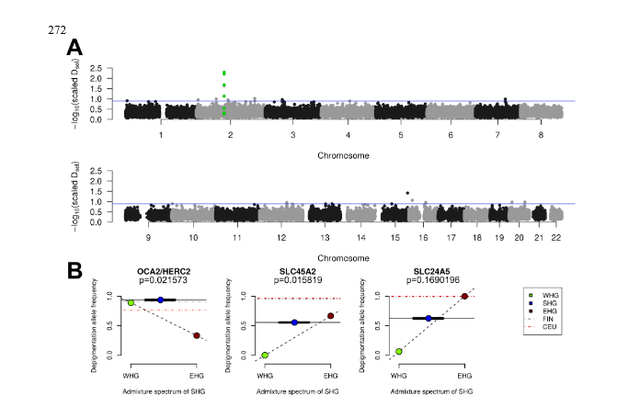 Figure 3: Adaptation to high-latitude climates By sequencing complete ancient genomes, we can compute unbiased estimates of genetic diversity, which are informative of past population sizes and population history. Here, we restrict the analysis to WHGs and SHGs, since only SNP capture data is available for EHGs (Supplementary Information 7). In current-day Europe, there is greater genetic diversity in the south compared to the north. During the Mesolithic, by contrast, we find higher levels of genetic diversity (Supplementary Information 7) as well as lower levels of runs of homozygosity (Fig. 2A) and linkage disequilibrium (Fig. 2B) in SHGs compared to WHGs (represented by Loschbour and Bichon, (15, 29)) and Caucasus hunter-gatherers (CHG, represented by Kotias and Satsurblia, (29)). Using a sequential-Markovian-coalescent approach (30) for the high-coverage, high quality genome of SF12, we find that right before the SF12 individual lived, the effective population size of SHGs was similar to that of WHGs (Fig. 2C). At the time of the LGM and back to c. 50,000 years ago, both the WHGs and SHGs go through a bottleneck, but the ancestors of SHGs retained a greater population size in contrast to the ancestors of WHGs who went through a more severe bottleneck (Fig. 2c). Around 50,000-70,000 years ago, the effective population sizes of the ancestors of SHGs, WHGs, Neolithic groups (represented by Stuttgart (15)) and Paleolithic Eurasians (represented by Ust-Ishim (31)) align, suggesting that these diverse groups all trace their ancestry back to a common ancestral group which likely represents the early migrants out-of-Africa, who likely share a common ancestry outside of Africa. 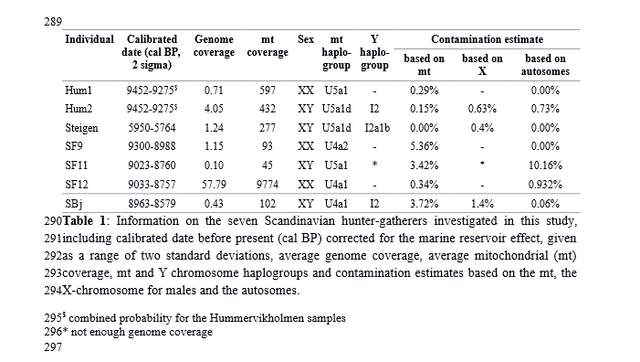 Adaptation to high-latitude environments With the aim of detecting signs of adaptation to high-latitude environments and selection during and after the Mesolithic, we employed three different approaches that utilize the Mesolithic genomic data. In the first approach, we assumed that SHGs adapted to high-latitude environments of low temperatures and seasonally low levels of light, and searched for gene variants that carried over to modern-day people in northern Europe. As we have already noted, modern-day northern Europeans trace limited amount of genetic material back to the SHGs (due to the many additional migrations during later periods), and any genomic region that displays extraordinary genetic continuity would be a strong candidate for adaptation in people living in northern Europe across time. We designed a statistic, D sel (Supplementary Information 10), that captures this specific signal and scanned the whole genome for gene-variants that show strong continuity (little differentiation) between SHGs and modern-day northern Europeans while exhibiting large differentiation to modern-day southern European populations (32) (Fig. 3A; Supplementary Information 10). Six of the top ten SNPs with greatest D sel values were located in the TMEM131 gene that has been found to be associated with physical performance (33), which could make it part of the physiological adaptation to cold (34). This genomic region was more than 200kbp long and showed the strongest haplotypic differentiation between modern-day Tuscans and Finns (Supplementary Information 10). The particular haplotype was relatively common in SHGs, it is even more common among today’s Finnish population (Supplementary Information 10), and showed a strong signal of local adaptation (Supplementary Information 10). Other top hits included genes associated with a wide range of metabolic, cardiovascular, developmental and psychological traits (Supplementary Information 10) potentially linked to physiological (34). 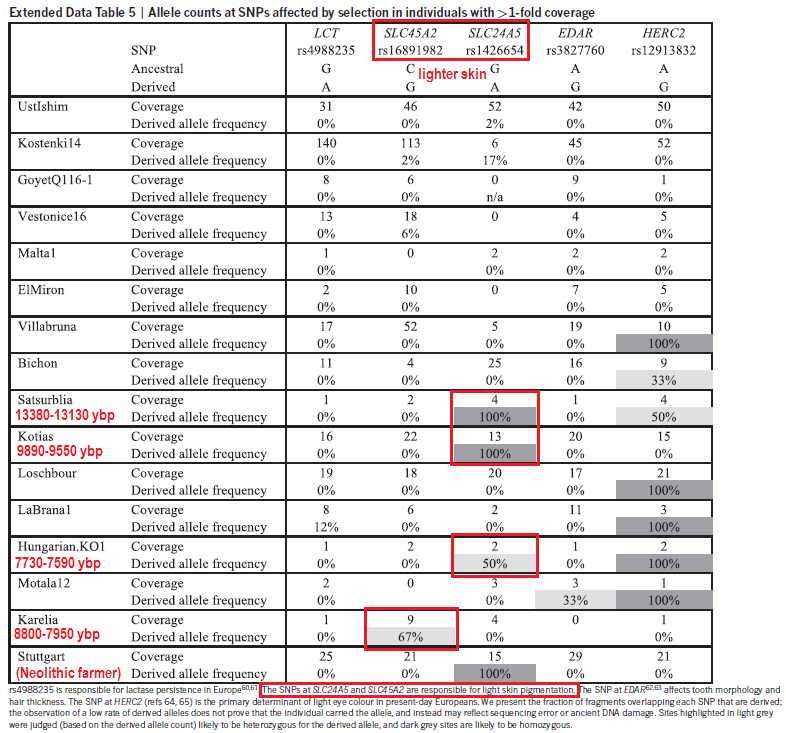 In addition to performing this genome-wide scan, we studied the allele frequencies in three pigmentation genes (SLC24A5, SLC45A2, having a strong effect on skin pigmentation, and OCA2/HERC2, having a strong effect on eye pigmentation) where the derived alleles are virtually fixed in northern Europeans today. The differences in allele frequencies of those three loci are among the highest between human populations, suggesting that selection was driving the differences in eye color, skin and hair pigmentation as part of the adaptation to different environments (35–37). The SHGs show a combination of eye and skin pigmentation that was unique in Mesolithic Europe, with light skin pigmentation and varied blue to light-brown eye color. This is strikingly different from the WHGs – who have been found to have the specific combination of blue-eyes and dark-skin (15, 17, 18) (Fig. 3B) – and EHGs – who have been suggested to be brown eyed and light-skinned (16, 17) (Fig. 3B). The unique configuration of the SHGs is not fully explained by the fact that SHGs are a mixture of EHGs and WHGs as the frequencies of the blue-eye and one light-skin variant are significantly higher in SHGs than expected from their genome-wide admixture proportions (Fig. 3B, Supplementary Information 10). This could be explained by a continued increase of the allele frequencies after the admixture event, likely caused by adaptation to high-latitude environments (35, 37). |
|
|
|
Post by Admin on Aug 13, 2017 19:32:05 GMT
Conclusion By combining information from climate modeling, archaeology and Mesolithic human genomes, we were able to reveal the complexity of the early colonization process of Scandinavia and human adaptation to high-latitude environments. We disentangled migration routes and linked them to particular archaeological patterns, demonstrate greater genetic diversity in northern Europe compared to southern Europe – in contrast to modern-day patterns – and show that many genetic variants that were common in the Mesolithic have been lost today. These finds reiterate the importance of human migration for dispersal of novel technology in human prehistory (13–16, 21, 27, 38–45) and the many partial population turnovers in our past. 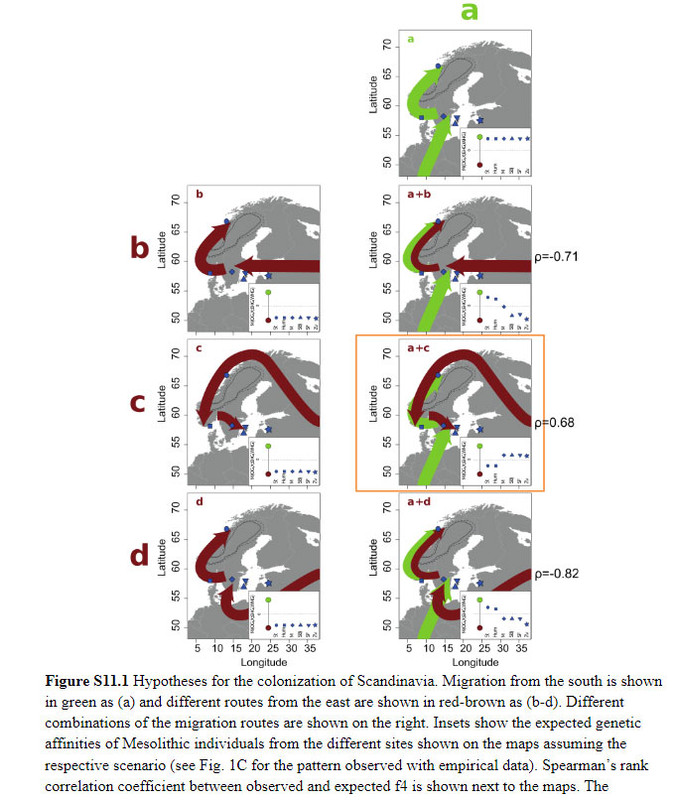 S11 Genetic testing of the post-glacial migration routes into Scandinavia The genomic affinities of the Mesolithic Scandinavians analyzed in this study suggest a complex migration pattern. Based on geography, genetics, the position of the ice-sheet on the Scandinavian peninsula and archaeology (see section S1, Figure S1.2), we hypothesize different migration routes into Scandinavia around 10,000 BP: a) a migration of WHGs from the south, b) a migration of EHGs from the east across the Baltic Sea, c) a migration of EHGs from the east and along the north-Atlantic coast, d) a migration of EHGs from the east and south of the Baltic Sea, as well as combinations of these four migration routes (Figure S11.1). These hypothetical scenarios allow us to formulate expectations of the genetic pattern seen in the different individuals. A migration of only WHGs would cause strong affinities of all SHGs to that group, whereas an exclusively EHG-like migration would make all SHGs very similar to EHGs. If there were combinations of migrations from both sources, a pattern of geographic structure would emerge: different SHGs would be characterized by differential affinities to WHGs and EHGs. We formulate these affinities in terms of expected tendencies for f4 (Chimp, X; EHG, WHG) which are shown in Figure S11.1. We only give qualitative expectations as a lot of additional information regarding population splits, drift parameters and admixture proportions would be required to formulate quantitative expectations for f4 . The qualitative expectations still allow us to test rank correlations between expected values and observed values of f4 (Chimp, X; EHG, WHG) to evaluate how the different hypotheses fit the data. 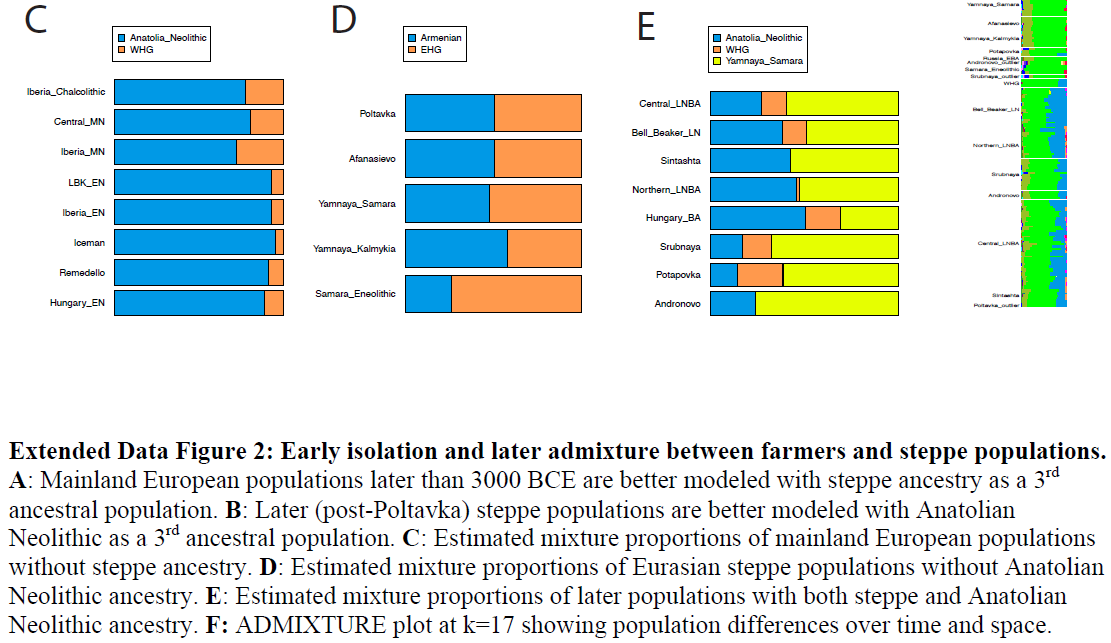 These rank correlations are not informative for single migrations but the data suggests that SHGs are in fact a mixture of EHGs and WHGs. The only scenario that produces a positive rank correlation (rho=0.677, p=0.004) between observed and expected values is a combination of (a) and (c) – one migration of WHGs from the south and another of EHGs from the east following the Norwegian coast. Such a scenario explains the higher affinities of Norwegian SHGs with EHGs as well as the higher affinities of Swedish SHGs with WHGs. The qpAdm admixture proportions (Figure 1A) are also significantly different between Norwegian and Swedish Mesolithic individuals (Wilcoxon test; p=0.01399). These affinities are consistently observed across all population genomic analyses in this study (PCA, ADMIXTURE, f4, qpAdm, TreeMix) and they are not correlated with the age of the samples (Figure S11.2). Therefore, we conclude that two migrations into Scandinavia fit the genetic data: a migration from central Europe into southern Sweden and a second migration along the Norwegian coast and around the ice sheet covering the center of the Scandinavian peninsula. The populations spread across Scandinavia and mixed creating the geographic population structure seen in the genomic data. |
|
|
|
Post by Admin on Oct 25, 2017 18:58:15 GMT
Genotypes under selection Imputation permitted us to follow the temporal dynamics of genetic variants that are believed to have been under selection. Of two skin pigmentation loci known to have swept to fixation during European prehistory32,33, the light pigmentary variant of SLC24A5 is present from the earliest of our samples and is homozygous from the Middle Neolithic onwards, whereas the light pigmentary variant of SLC45A2 only appears towards the later half of our transect with the first homozygote genotype in the Copper Age (Fig. 3). Both SLC24A5 and SLC45A2 exhibited an ancestral homozygous state in Mesolithic specimens of Central5 and Western Europe7, while SLC24A5 had the derived state in a Central European Neolithic individual5. Our temporal transect suggests separate selective sweeps at these two pigmentary loci, acting over a millennium apart. The selected variant at a third pigmentary locus with a proposed adaptive history in Europe, TYRP1, also shows some tendency to higher prevalence in later samples. This temporal transition towards lighter pigmentation is also seen with hair where colours and shades estimated from SNPs used in the forensic Hirisplex system grade from black/dark brown in earlier samples to light brown and dark blonde in later individuals (Fig. 3). One of the strongest signals of selection within human genome variation is that around the lactase persistence allele in Europeans; a response to a dietary focus on raw milk from domestic cattle. It has been postulated that this allele first underwent selection 5,500 years BC, possibly in association with the Neolithic LBK culture within Central Europe34. Here in our temporal sequence, its appearance is delayed until the more recent of our Bronze Age individuals, who lived only ~1,000 years BC.  Beyond inferences about individual phenotypes, we have used our results to examine the population genetic affinities of a temporal transect of genome sequences from burials on the Great Hungarian Plain, a region of high archaeological significance for major European cultural transitions. We investigated samples across a diversity of archaeological cultures and show evidence for major shifts in genome affinity accompanying the advents of the Neolithic, Bronze and Iron Ages, strongly implying that these changes in material culture were accompanied by substantial migrations. The Neolithic genomes reported here accord with prior German, Scandinavian and Alpine early farmer genomes in showing an immigrant signature of Southern Mediterranean affinity2,5,6,8. However, an intriguing finding is that of a single individual with a strongly Mesolithic genomic signature within the context of the Körös culture, part of the earliest Neolithic of Southern Europe. This is the earliest genetic indication of contact between these two subsistence strategies. In the Middle and Late Hungarian Neolithic local Mesolithic influence is further discernible through the appearance of mtDNA and Y-chromosome haplogroups typical of European hunter-gatherer populations, concurring with other evidence for admixture in the ancestry of European farmers5,8,22,23. Similar to the Tyrolean Copper Age iceman6 our Copper Age (Baden Culture) sample shows similarity to Neolithic genomes, in accordance with archaeological continuity in the region. In contrast, the Bronze Age genomes shift towards an affinity to Central Europe, suggesting migratory influence from the North. The single pre-Scythian IR1 genome shows another shift towards migration from the East. Altogether, our results accord with archaeological perspectives that link these major transitions in European material culture to population movements rather than cultural diffusion alone. Nature Communications 5, Article number: 5257 (2014) |
|
|
|
Post by Admin on Nov 21, 2017 19:08:53 GMT
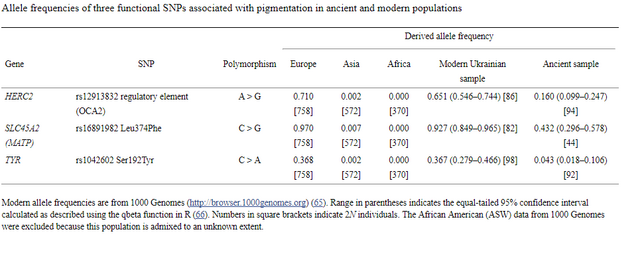 Table 1. Allele frequencies of three functional SNPs associated with pigmentation in ancient and modern populations Genomic signatures of natural selection in humans are usually obtained from modern population genetic data and take the form of patterns of variation outside those expected under neutrality (1), including strong correlations between allele frequencies and hypothesized ecological drivers of selection (2) and identifying alleles with unusually recent age estimates for their frequencies (1). All such indirect approaches have poor sensitivity and temporal resolution, most are confounded by past demographic processes, and many are insensitive to selection acting on standing variation (3). With advances in ancient DNA analysis techniques it is possible to obtain direct estimates of natural selection over specific time periods by estimating allele frequency change, permitting changes in selection intensity to be detected through time and a more detailed understanding of the forces shaping human evolution. However, to date no such estimates have been made. Pigmentation is a particularly conspicuous human phenotypic variation and in the past has been misleadingly used as a proxy for deep biogeographical origins (4). Dark pigmentation is thought to be the ancestral state in humans and to have been maintained by purifying selection in low-latitude, high-UVR regions to protect against folate photolysis, UV radiation (UVR)-induced DNA damage, and possibly damage to immunoglobulins (5, 6). Continental-scale correlations between skin pigmentation and incident UVR levels strongly indicate positive ecological adaptation (5), although sexual selection—particularly in relation to eye and hair coloration (7)—and relaxation of selective constraints (6) may also have been important. 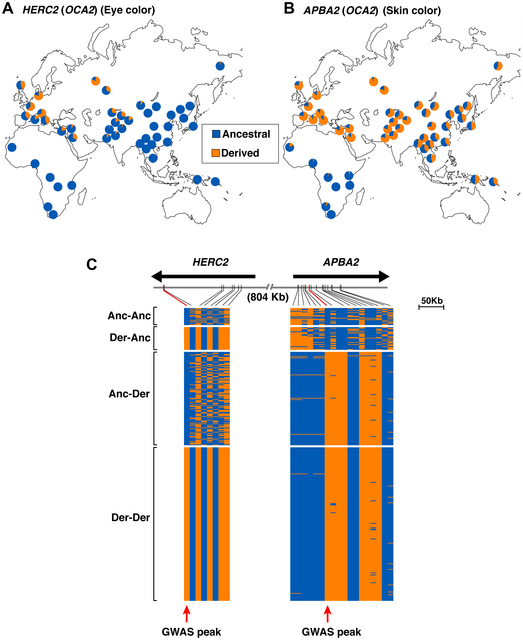 Melanin, a derivative of tyrosine, is the primary biopolymer responsible for constitutive animal pigmentation and is found in two forms in humans, eumelanin (black-brown) and pheomelanin (red-yellow). It is synthesized in melanosomes, organelles located in melanocytes in several tissues including the basal layer of the epidermis, hair follicles, and the iris. Variation in pigmentation depends mainly on differences in the amount and type of melanin synthesized and the shape and distribution of melanosomes in different tissues (8). The products of several genes are involved in melanin synthesis and distribution, and known DNA polymorphism in those genes explains a substantial proportion of human pigmentation variation (9–12). Signatures of recent natural selection have been detected in some pigmentation genes, including both shared and regionally specific alleles associated with lighter skin pigmentation in eastern and western Eurasia (13, 14), using modern population genetic data (1, 12, 13, 15, 16). To obtain direct estimates of the strength of natural selection driving depigmentation we analyzed three polymorphic sites in ancient and modern samples (Table 1) that had previously been identified through genome-wide association studies (GWAS) (1, 17) and fine-mapping SNP association (18) as influencing pigmentation in modern Europeans: HERC2 (rs12913832 A > G) (18), SLC45A2 (rs16891982 C > G) (13), and TYR (rs1042602 C > A) (11). The product of the TYR gene, tyrosinase, catalyzes the first two steps of the melanogenesis pathway (8), and its absence generates an epistatic mask on downstream pigment-coding genes, halting melanin production. The TYR SNP rs1042602 is highly polymorphic in Europeans, and the derived A allele has been associated with light skin (19) and eye color (20) and the absence of freckles (11). SLC45A2 (MATP) is involved in the distribution and intracellular processing of tyrosinase and other pigmentation enzymes (21). The derived rs16891982 G allele, which decreases in frequency along a north–south cline in Europe (13), is associated with lighter skin, hair, and eye pigmentation in modern populations (13, 22). The HERC2 SNP rs12913832 A > G is the main determinant of iris pigmentation (brown/blue) (18, 23) and is also associated with skin and hair pigmentation and the propensity to tan (24). It is located within an intron of the Hect Domain and RCC1-like Domain2 (HERC2) gene 21 kb upstream from the OCA2 promoter and serves as an enhancer for OCA2 expression (23, 25). OCA2 expression is increased in the melanocytes of carriers of the ancestral A allele, and attenuated in carriers of the derived G allele (25). OCA2 encodes a melanosomal transmembrane protein, protein P, which is involved in the trafficking and processing of tyrosinase, the regulation of melanosomal pH, and glutathione metabolism (26). Selection favoring the SLC45A2 rs16891982 G allele has been estimated to have begun between 11,000 and 19,000 y ago (14), well after the expansion of anatomically modern humans out-of-Africa. The age of the derived TYR rs1042602 allele has been estimated using two different methods: rho statistics place its emergence around 6,100 y ago, whereas the Bayesian coalescent approach using BEAST dates the allele to ∼15,600 y ago (27). |
|
|
|
Post by Admin on Nov 23, 2017 19:21:19 GMT
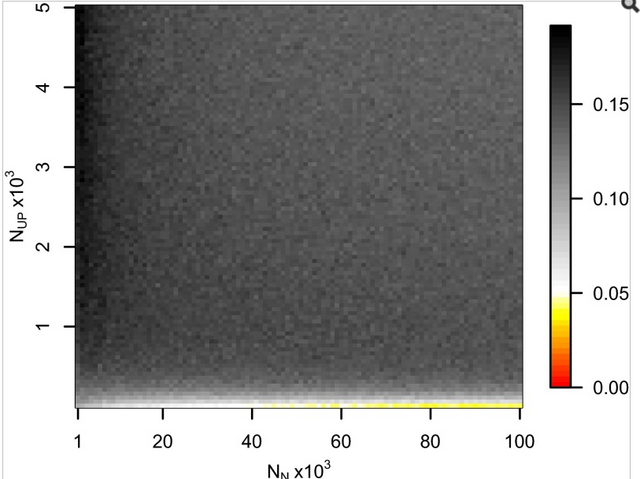 Fig. 1. Probabilities of obtaining FST equal to or greater than that observed (0.00551) between 60 Eneolithic (ca. 6,500–5,000 y ago) and Bronze Age (ca. 5,000–4,000 y ago) samples from the Pontic–Caspian steppe, and a combined sample ... Ancient DNA was retrieved from 63 out of 150 Eneolithic (ca. 6,500–5,000 y ago) and Bronze Age (ca. 5,000–4,000 y ago) samples from the Pontic–Caspian steppe, mainly from modern-day Ukraine. We used multiplex-PCR enrichment and next-generation sequencing to genotype the three pigmentation-associated SNPs (rs12913832, rs16891982, and rs1042602) and mtDNA hypervariable region 1 (HVR1) sequences plus 32 mtDNA coding region SNPs and a 9-bp-indel from these individuals (Tables S1 and S2). Consensus HVR1 sequences were successfully assembled from 60 individuals. Pigmentation gene data were obtained from 48 samples. We also genotyped the three pigmentation-associated SNPs in a sample of 60 modern Ukrainians (28) and observed an increase in frequency of all derived alleles between the ancient and modern samples from the same geographic region (Table 1 and Fig. S1). This implies that the pigmentation of the prehistoric population is likely to have differed from that of modern humans living in the same area. Modern frequencies of the derived alleles within all of Europe and outside of Europe are provided for comparison (Table 1). Inferring natural selection based on temporal differences in allele frequency requires the assumption of population continuity. To this end we compared the 60 mtDNA HVR1 sequences obtained from our ancient sample to 246 homologous modern sequences (29–31) from the same geographic region and found low genetic differentiation (FST = 0.00551; P = 0.0663) (32). Coalescent simulations based on the mtDNA data, accommodating uncertainty in the ancient sample age, failed to reject population continuity under a wide range of assumed ancestral population size combinations (Fig. 1). 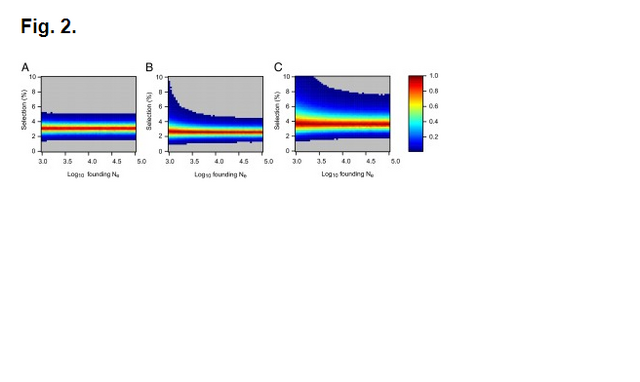 Conversely, continuity between early central European farmers and modern Europeans has been rejected in a previous study (33). However, the Eneolithic and Bronze Age sequences presented here are ∼500–2,000 y younger than the early Neolithic and belong to lineages identified both in early farmers and late hunter–gatherers from central Europe (33). A plausible explanation for this is that the prehistoric populations sampled in this study are a product of admixture between in situ hunter–gatherers and immigrant early farmers during the centuries after the arrival of farming, and that this admixture was a major process shaping modern patterns of mtDNA variation (34) and possibly also the variability observed in European hair, eye, and skin color. To test whether the observed increases in the three light pigmentation-associated alleles can be explained by genetic drift alone or whether natural selection needs to be invoked, we performed forward computer simulations of drift plus selection, accommodating uncertainty in ancient and modern allele frequency, population size, and ancient sample age. We assumed codominance for both SLC45A2 rs16891982 G and TYR rs1042602 A alleles (22, 35) and that the derived HERC2 rs12913832 G allele is recessive (36). Using these simulations, neutrality (S = 0) was rejected under all assumed ancestral effective population sizes—ranging from 103 to 105 at the time of the ancient sample (SLC45A2 P < 1 × 10−5, TYR P < 2 × 10−5, and HERC2 P < 1 × 10−5). The values of selection acting on the SLC45A2 rs16891982 G allele, the TYR rs1042602 A allele, and the HERC2 rs12913832 G allele that best explained the observed derived allele frequency changes were 0.030, 0.026, and 0.036, respectively (Fig. 2). Whereas there is strong evidence that the derived HERC2 rs12913832 G allele is recessive (36), it is less clear whether the SLC45A2 and TYR derived alleles are codominant, recessive, or dominant (22, 35). Under the assumption that both SLC45A2 and TYR derived alleles are recessive, the selection values that best explain the observed changes in frequency are 0.022 and 0.104, respectively, and under the assumption that they are dominant the selection values are 0.088 and 0.016, respectively; again, neutrality was rejected for all three alleles (P < 4 × 10−5) under all ancestral population sizes modeled and all assumptions of dominance/codominance/recessivity (Fig. S2). |
|

