|
|
Post by Admin on Feb 26, 2016 1:40:05 GMT
 The Yakuts (or Sakhas) from the Republic of Sakha are of particular interest because they contrast with other populations from Siberia in many respects such as in their specific funeral practices. Before Christianization, part of the population was buried, which is exceptional in this region of the world as most bodies were put in aerial graves on platforms or on trees. At the same time, the Yakuts are considered as the most remarkable example of northward expansion into Siberia [1] and especially as these semi-nomadic herders contrast so strikingly with surrounding Siberian populations on the basis of their Turkic language and their horse- and cattle-breeding economy. The geographic specificities of Central Yakutia, favorable to the development of a pastoralist economy and reflecting the environmental conditions of the South, certainly initiated the development of the Yakut culture in this region. Although several hypotheses regarding the geographic origin of this atypical population have been proposed from results of the first archaeological investigations of AP Okladnikov [2] to the latest population genetic studies [3-6], the precise origins of the Yakuts and their admixture with the indigenous tribes of Siberia, especially the Tungus, have been debated. Nevertheless, archaeological data currently agree with the appearance of the Yakut culture around the 14th century A.D. [7]. Recent population genetics data tend to support a dual origin for the maternal lineages of the Yakuts and admixture with the Tungus, and demonstrate that the paternal lines observed today result from a strong bottleneck that led to the restriction of these lineages in the Yakut population [3,4,6]. This does not, however, address the question of the number of settlers and the precise origin of the paternal lineages, or the admixture rate with indigenous populations [6]. Furthermore, the demographic changes occurring in the Yakut population since the Russian colonization make it difficult to reconstruct the genetic history using present day samples. Russians came into contact with the Yakuts in 1632. Christianization began around 1760 and by the beginning of the 19th century, a Christian ritual was performed in the majority of burials. From the 17th to the 19th century smallpox and measles epidemics, caused by contact with Russians settlers, decimated the Yakut people and their neighbors, and during the Second World War a large proportion of young Yakut males was killed. Therefore ancient DNA (aDNA), which allows the direct study of an ancient population without their descendants, represents a major advantage in order to avoid the possible bias represented by recent genetic events [8,9]. 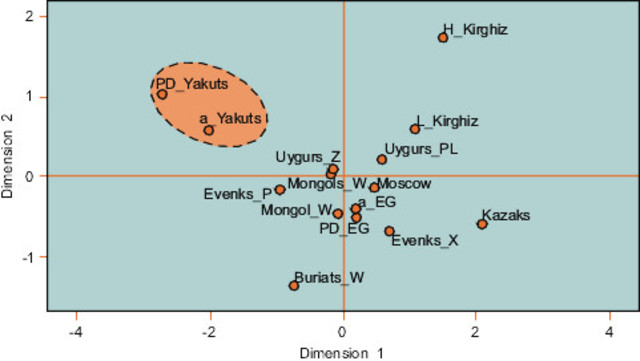 MDS plot of genetic distances between populations on the basis of Y- STRs. Sixty one percent (8 out of 13) of the haplotypes (Ht1, Ht2, Yaka56, 65, 71, 80, 81, 86) were affiliated to the N1c (TAT-C) haplogroup on the basis of the SNP analyses. This haplogroup is considered as the most frequent in the Yakut population, and its frequency varies across studies from 75% [12] to 100% [13]. Sample YAKa26 was affiliated to haplogroups K. The SNP typing was inconclusive for 5 individuals (YAKa17, 19, 47, 49 and 57); nevertheless the affiliation to N1c was excluded on the basis of the absence of the TAT-C mutation. Although all the Y-STR haplotypes have been transmitted to the present day Yakuts, only a few of them are shared with other populations. A comparison to the YHRD database and other literature data showed that only Ht1 and the haplotype of the YAKa26 individual matched with Evenk male lineages; confirming the specificity of the N1c lineages. In this case, one can assume that N1c comes from Yakut ancestors considering the low N1c frequency in Evenks [6,14]. Moreover, the ethnical affiliation of one individual to either the Yakut or the Evenk population could be difficult nowadays, considering that these two ethnic groups share a similar lifestyle or language in some areas of Yakutia. The Y-STR haplotypes of YAKa17 and YAKa26 were found in present day Mongols, Buryats, Kalmyks and Central Asians. 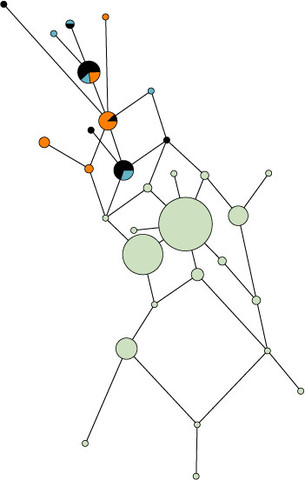 Median Joining network based on Y-chromosomal haplotype. Median Joining network based on Y-chromosomal haplotypes belonging to the haplogroup N1c. Black: ancient Yakuts, Blue: present day Yakuts, Orange: Evenks. Green for Other populations included in the computations: Buryats [75,80], Evenks [80,81], Mongols [76,80], Tuvans [43,80], Uyghurs [76,80] and Altaians, Evens, Kalmyks, Khakassians, Koryaks, Shors, Sojots, Tofalars, and Yakuts from Derenko et al. [80]. The origin of the most frequent Y-chromosomal haplotypes (Ht1 and Ht2) was difficult to establish on the basis of genetic information. Indeed, these two lineages belonging to haplogroup N1c seem to be restricted to Yakut populations, and were probably present since the period they were first located in Central Yakutia. Interestingly, the comparison with archaeological data revealed that the male individuals (YAKa34, 39, 40, 69, 78) at the beginning of the 18th century, identified as Clan Chiefs (or tojons) on the basis of their grave goods (weapons, jewelry, silk clothes, richly ornamented saddles and signet rings), belonged to these two haplotypes. Therefore, archaeological data could bring interesting information in tracing back the origin of these enigmatic male lineages. Indeed, the grave goods of the 15th/17th centuries (weapons and horse harnesses) and the construction of coffins with an empty trunk from the 18th century are similar to the burial customs of the Cis-Baïkal area [44] and of the Egyin Gol Necropolis during the 3rd century BC [45-47]. This suggests that the male ancestors of the Yakuts were probably formed of a small group of horse-riders originating from Northern Mongolia or the Baïkal Lake. Later, the patrilineal clan organization of the Yakuts would have facilitated the diffusion of male lineages borne by the individuals of high social rank. This hypothesis is supported by the fact that the Y-chromosomal haplotypes identified by the warriors of the 15th/17th centuries were transmitted to the tojons of the 18th century. After Russian colonization, the influence of the tojons was strengthened by the decrees made by the Empress Anna Ivanovna [1], and this official reinforcement of their power might have increased the spread of a limited number of Y-chromosomal lineages for a second time. Therefore, the early founder effect combined with the Yakutian traditions and the Russian influence would have led to the present day genetic pattern observed for the paternal lineages. Conversely, the results obtained from the mtDNA analyses revealed a more important diversity and varying origins of the maternal lineages. Indeed, the gene diversity observed in our ancient sample is intermediate compared with the diversity found in small and isolated Siberian groups such as the Chukchi and Mongolian [15,48], Buryatian [4] or Central Asian [23] populations. The haplogroup distribution observed in our sample matched that found in present day Yakuts and is constituted by distant haplogroups found in Siberian, Mongolian and Central Asian populations. The major haplogroup, C4, indicates more specific affinities with the Evenks [21]. Nevertheless, the individual comparison of maternal lineages with data from literature allowed us to precise that four out of the six lineages belonging to the C4 haplogroup were present in the Buryat population. Therefore, the Buryat origin of some of the D5 sub-haplogroups found in the Yakuts as demonstrated by Pakendorf et al. [6] could be expanded to the C4 haplogroup. The influence of both Buryat and Evenk populations is clearly visible in the mtDNA lineages of the ancient Yakuts. Concerning the contribution of the Evenks to the Yakut's mtDNA gene pool, the admixture might have mostly occurred between Yakut men and Evenk women (as assumed by Pakendorf et al. [6]) according to the high frequency of the C4 haplogroup in these two groups, and since the Y-chromosomal lineages are highly specific to the Yakuts and the genetic diversity indices are lower for Y chromosome than for mtDNA. Moreover, the number of first settlers who arrived in Central Yakutia was certainly limited and the patrilocal exogamy practiced by the traditional Yakut society [49] corroborates the inclusion of autochthonous women. This assumption is further confirmed by linguistic data that reveals intermarriages between the Yakuts and Evenks [50].  Finally, the stability through time demonstrated by the Y chromosome is also observed in the maternal lineages. Indeed, most of the sequences present in our ancient sample (83%) have been transmitted to the present day population. However, this could signal a small loss in variation over the two last centuries, which could be associated with stochastic processes linked to demographic changes undergone by the Yakut population (plague, smallpox) or other phenomena [51-53]. Besides, the Yakuts from all chronological periods are grouped together in the MDS plot and the Fst calculation revealed the absence of significant distances between them. Thus, even if the Russian colonization had an important impact on the expansion of the Yakuts throughout Yakutia [1], its genetic influence appears to be relatively low. BMC Evol Biol. 2010; 10: 25. doi: 10.1186/1471-2148-10-25 |
|
|
|
Post by Admin on Mar 5, 2016 1:20:06 GMT
There is great diversity in primates with regard to hair appearance, including its distribution, shape and colour1. Hair plays a range of important functions, including thermal regulation, camouflage, sensory and social signalling, and the evolution of hair has been proposed to be influenced by both natural and sexual selection1. Humans differ from other primates in having lost most terminal body hair (via hair follicle miniaturization), possibly in connection to the adaptive development of more efficient sweating, linked to bipedalism2, 3. However, considerable head hair has been retained in modern humans and its appearance shows extensive variation between individuals4, 5, 6. Head hair appearance is highly heritable7, 8, 9, 10 and certain traits show high differentiation between continental native populations. For instance, variation in hair colour is essentially restricted to West Eurasia11, whereas straight hair is virtually absent from sub-Saharan Africa4. It has been proposed that variation in head hair appearance has been influenced by selection during the evolution of modern humans2, 3, 11. Although genetic association studies for human hair traits are few to date, loci influencing male pattern baldness, scalp hair colour and shape (curliness) have been identified in samples of European and East Asian ancestry6. Consistent with the key role of androgens in balding, the androgen receptor (AR) gene region has been highlighted as a major determinant among several loci associated with male-pattern baldness12. The genes associated with hair colour, involved in various aspects of melanocyte biology and melanin synthesis, also often impact on skin and eye pigmentation13. Interestingly, different genes have been associated with straight hair in Europeans and East Asians, suggesting that this trait evolved independently at least twice. The most robust associations for straight hair have implicated Trichohyalin (TCHH, a structural hair protein) in Europeans14, 15, and EDAR (a cell signalling receptor) in East Asians16, illustrating the range of cellular mechanisms that can impact on hair shape.  At the top are shown drawings illustrating the seven hair features examined in the CANDELA study sample. Thick lines connect these features with the candidate genes identified in regions with SNPs reaching genome-wide significant association (Table 1). Several of the regions showing genome-wide significant association include strong candidate genes. Scalp hair shape and beard thickness are strongly associated with SNPs in 2q12 (Fig 3a,b) and two other traits (eyebrow thickness and monobrow) show genome-wide suggestive P values for SNPs in this same region (Supplementary Fig. 6). Associated SNPs overlap the EDAR (ectodysplasin A receptor) gene. EDAR acts as part of the EDA-EDAR-EDARADD signalling pathway23 during prenatal development to specify the location, size and shape of ectodermal appendages, such as hair follicles, teeth and glands23. Strongest association with hair shape was observed for SNP rs3827760 (Fig. 3a and Table 1), coding for a V370A substitution in EDAR. This variant has been robustly associated with hair shape in East Asians16, 24, 25. In a previous study of the CANDELA sample, which examined 30 ancestry informative markers, we found association between hair shape and a SNP in EDAR that is in strong linkage disequilibrium (LD) with rs3827760 in the 1000 genomes data17. The EDAR SNP showing strongest association with beard density is not the coding SNP rs3827760 associated with hair shape but rather rs365060 located ~62 kb upstream of rs3827760 in the first intron of EDAR (Fig. 3b). Several SNPs in this intron have smaller association P values than rs3827760 and analyses conditioning on rs3827760 suggest that the association signal of SNPs in this intron is independent from that observed at rs3827760 (Supplementary Fig. 7). These intronic SNPs are located in a different LD block and there is a recombination hotspot between them and rs3827760 (Fig 3a,b and Supplementary Fig. 7A). Interestingly, the first intron of EDAR is rich in regulatory elements and SNPs in this intron show evidence of recent selection in Europeans (discussed further below). Hypohidrotic ectodermal dysplasia is a Mendelian disorder caused by mutations in the EDA-EDAR-EDARADD pathway and is characterized by sparse scalp hair, eyebrows and eye lashes23. Transgenic mice with increased Edar function have been shown to have thickened and straightened hair fibres26, 27. We therefore examined chin hair follicle density in wild-type mice and in an Edar gain-of-function transgenic strain (EdarTg951/Tg951)26. Consistent with the effect of EDAR on beard thickness, we found that the EdarTg951/Tg951 strain has significantly lower chin hair follicle density compared with wild-type mice (Fig. 4a,b).  Figure 3: Association plots for six regions with SNPs showing genome-wide significant association to hair traits. Eyebrow thickness shows genome-wide significant association to SNPs on 3q23 overlapping the forkhead box L2 (FOXL2) gene (Fig. 3d). Rare mutations in the FOXL2 gene region (including coding variants as well as upstream and downstream intergenic rearrangements) cause blepharophimosis syndrome (BPES)34, an autosomal dominant eyelid malformation often accompanied by thick eyebrows. Mouse experiments have shown that Foxl2 is expressed around the eyes up to the time that hair is formed (E13.5)35 and a mutant with altered Foxl2 expression (and BPES features) typically shows hair loss around the eyes36. Monobrow shows genome-wide significant association with SNPs in 2q36 with strongest association being observed for marker rs2395845 located ~70 kb downstream of the paired box gene 3 (PAX3) gene, an interesting candidate in the region (Fig. 3e). Rare mutations of PAX3 have been shown to cause Waardenburg syndrome type 1 (WS1). WS is a clinically and genetically heterogeneous Mendelian disorder of neural crest derivatives whose manifestations include deafness, a range of pigmentation abnormalities, broad nasal bridge and monobrow (seen in ~85% of WS1 patients37). Intronic SNPs within PAX3 have been implicated in recent GWASs of facial morphology, particularly in relation with nasion position (the point just above the nasal bridge)38, 39. PAX3 is a key transcription factor during embryogenesis and analysis of mouse mutants have confirmed that it is essential to guide normal development of neural crest derivatives40.  Figure 4: EDAR effects on mouse facial hair follicle density and expression of PRSS53 in anagen (growing) human hair follicles. Hair greying shows genome-wide significant association to SNP rs12203592 in intron 4 of the interferon regulatory factor 4 gene (IRF4; Fig. 3f). This SNP also shows association with hair colour in our sample (Table 1) and in previous studies this SNP has been associated with skin, hair and eye pigmentation13. Recent in-vitro studies have shown that rs12203592 impacts on the function of an enhancer element regulating IRF4 expression and the induction of tyrosinase (TYR), a key enzyme in melanin synthesis41. In addition to IRF4, hair colour (but not hair greying) shows genome-wide significant association to four other well-established pigmentation gene regions (SLC45A2, TYR, OCA2/HERC2 and SLC24A5; Table 1 and Supplementary Fig. 8)13. Finally, for balding, we replicate association to the well-established Androgen Receptor/Ectodysplasin A2 Receptor (AR/EDA2R) locus on Xq12 (ref. 12; Supplementary Fig. 8). The other four genomic regions showing genome-wide association include no strong candidate genes with established roles in hair biology (Table 1 and Supplementary Fig. 8). Beard thickness is associated with SNPs in 7q31, 4q12 and 6q21 where the nearest genes are, respectively: forkhead box P2 (FOXP2), ligand of numb-protein X 1 (LNX1) and prolyl endopeptidase (PREP; Supplementary Fig. 8). None of these genes have known functions specifically related to hair. Similarly, balding is associated with SNPs in the second intron of the Glutamate receptor delta-1 subunit gene (GRID1) on 10q22 but this gene has no documented role in hair biology.  Figure 5: Processing of PRSS53 and signals of selection in the PRSS53 gene region. The colour of hair results from melanin pigments transferred to hair fibre keratinocytes from hair follicle melanocytes. These melanocytes differentiate from melanoblasts that migrate from the neural crest into hair follicles early in development6. Some hair follicle melanoblasts remain undifferentiated and serve as stem cells for the periodic replenishment of mature melanocytes, melanogenesis occurring only in the anagen phase of the hair growth cycle. Among the several hundred gene products known to participate in melanogenesis, recent association studies have identified a handful that influence hair colour variation in Europeans13. Most of these associations have been replicated here (Table 1), including that of the derived T allele at SNP rs12203592 in IRF4 with lighter hair colour. It has been shown experimentally that IRF4 interacts with the microphthalmia-associated transcription factor (MITF, a key regulator of the expression of many pigment enzymes and differentiation factors), to activate the expression of TYR (a rate-limiting essential enzyme in melanin synthesis). The derived T allele at rs12203592 leads to reduced TYR expression and melanin synthesis, consistent with the association of this allele with lighter hair colour41. In line with the geographic distribution of light hair colour, the T allele at rs12203592 is essentially absent outside Europe (Supplementary Table 7). Interestingly, we find that the T allele at SNP rs12203592 is also associated with increased hair greying. Experimental evidence suggests that the mechanism of hair greying involves incomplete maintenance of melanocyte stem cells in the hair follicle6. Importantly, MITF is known to affect melanocyte survival via its regulation of anti-apoptotic Bcl2 expression, a key factor in protection of the hair follicle against oxidative stress6. To probe the mechanism by which IRF4 might impact on hair greying, it will therefore be important to evaluate whether the T allele at rs12203592 influences MITF in terms of melanoblast stem cell maintenance and survival or via melanocyte loss post differentiation. The skin on different parts of the body has different hair characteristics, with the final hair distribution dependent upon the spacing pattern laid down during development, the extent of skin growth that occurs subsequent to pattern establishment, and to hormonal and ageing effects. The development of skin at different body sites is known to be controlled by an underlying transcription factor code52 to which FOXL2, FOXP2 and PAX3 may contribute in defining hair distribution on specific areas of the face. As hair of the beard is produced through a two-stage process of embryonic hair follicle patterning followed by a post-pubertal androgen-driven transformation into terminal hair, beard thickness could be modulated by genes acting either prenatally or at puberty. Our finding of reduced hair placode density in embryonic mice with increased Edar expression suggests that the basis for this variation lies in the recognized role of EDAR in developmental hair patterning53. It is likely that EDAR function affects hair follicle density on most or all of the human body, as described in the mouse26, but that on the head this effect is most readily apparent as variation in beard thickness. Analyses focusing on the mechanism by which associated genetic variants affect regional facial hair density should provide insights into developmental patterning in humans, and perhaps yield clues into the genetic basis for the striking modification of hair distribution that has occurred in human evolution54. Adhikari, K. et al. A genome-wide association scan in admixed Latin Americans identifies loci influencing facial and scalp hair features. Nat. Commun. 7:10815 doi: 10.1038/ncomms10815 (2016). |
|
|
|
Post by Admin on Mar 9, 2016 1:29:16 GMT
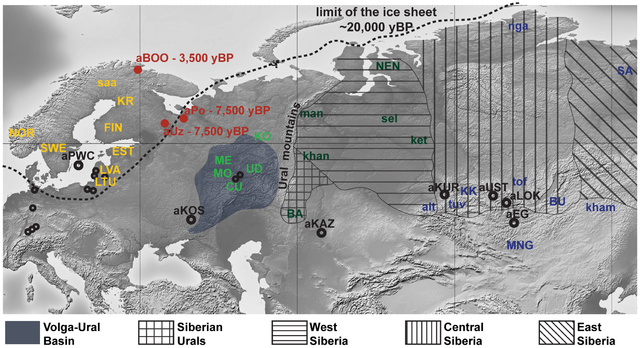 Figure 1. Map of Eurasia showing the approximate location of ancient (uncalibrated dates) and present-day Eurasian samples. The absence of strong structure in the present-day mtDNA gene pool of NEE stands in contrast to the variety of languages and cultures, and to the complex history of how and when these were formed. Modern mtDNA data does not resolve the origins of the Saami either. Our aim was to provide answers to these questions and reconstruct events in the genetic history of NEE by generating and analyzing ancient DNA (aDNA) data from prehistoric human remains collected in northwest Russia (Figure 1). In particular, our objective was to characterize the genetic relationships between hunter-gatherer populations in NEE and Central/Northern Europe and to estimate the genetic legacy of ancient populations to present-day NEE and Saami. The oldest samples were collected in the Mesolithic graveyards of Yuzhnyy Oleni Ostrov (aUz; ‘Southern Reindeer Island’ in Russian) and Popovo (aPo), both dated around 7,000–7,500 uncalibrated. yBP, uncal. yBP. The sites of aUz and aPo are located along one of the proposed eastern routes for the introduction of Saami-specific mtDNA lineages [32]. Results from odontometric analyses suggested a direct genetic continuity between the Mesolithic population of Yuzhnyy Oleni Ostrov and present-day Saami [41]. We also analyzed human remains from 3,500 uncal. yBP site Bol'shoy Oleni Ostrov (aBOO; ‘Great Reindeer island’ in Russian) in the Kola Peninsula. This site is located within the area currently inhabited by the Saami. We compared the ancient mtDNA data from NEE with a large dataset of ancient and modern-day Eurasian populations to search for evidence of past demographic events and temporal patterns of genetic continuity and discontinuity in Europe. 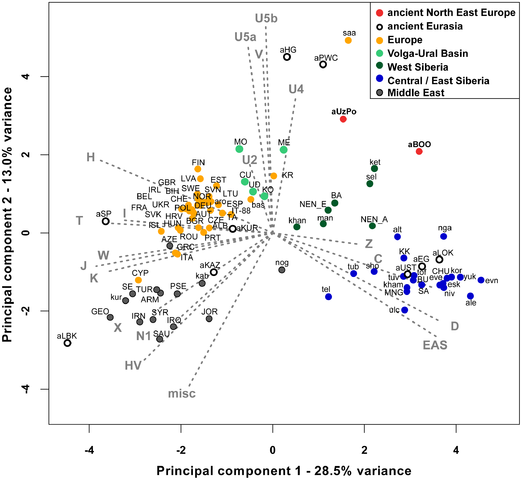 Figure 2. Principal Component Analysis of mitochondrial haplogroup frequencies. In order to identify the genetic affinities of the two ancient populations with other ancient and present-day Eurasian populations, mtDNA hg distributions were compared by Principal Component Analysis (PCA). The PCA plot of the first two components (41.5% of the total variance, Figure 2) showed that present-day populations largely segregate into three main clusters: Europeans (in yellow), Middle Easterners (in grey) and Central/East Siberians (in blue). The spread of extant populations of Europe and Central/East Siberia along the first component axis (28.5% of the variance) appeared to reflect their longitudinal position, whereas Europeans and Middle Easterners were separated along the second component axis (13.0% of the variance). As shown previously, populations of the ‘Central/East Siberian’ cluster were predominantly composed of hgs A, B, C, D, F, G, Y, and Z, while in contrast populations of the ‘European’ cluster were characterized by higher frequencies of hgs H, HV, V, U, K, J, T, W, X, and I (e.g., [43]–[47]). The two ancient groups - aUzPo and aBOO - from two individual time periods appeared remarkably distinct on the basis of the PCA, suggesting a major genetic discontinuity in space and time. The hg distribution in the Mesolithic aUzPo population: U4 (37%), C (27%), U2e (18%), U5a (9%), and H (9%), indicated an ‘admixed’ composition of ‘European’ (U4, U2e, U5a and H, 73%) and ‘Central/East Siberian’ (C, 27%) hgs, based on the PCA plot (Figure 2). Interestingly, the population of aUzPo did not group with modern NEE populations, including Saami, but fell instead between the present-day ‘European’ and ‘Central/East Siberian’ clusters on the PCA graph, and more precisely between populations of the VUB (in light green) and West Siberia (in dark green). The high frequency of hg U4 is a feature shared between Mesolithic aUzPo, present-day VUB (Komi, Chuvashes, Mari), and West Siberian populations (Kets, Selkups, Mansi, Khants, Nenets), with the latter group also being characterized, like aUzPo, by the presence of hg C. The genetic affinity between Mesolithic aUzPo and present-day West Siberian populations could be visualized on the genetic distance map of North Eurasia (Figure 3A), on which locally lighter colorings indicated low values of genetic distances, and therefore an affinity between aUzPo and extant West Siberians. In order to test the potential population affinities formulated on the basis of the hg-frequency PCA and the distance map, we examined the present-day geographical distribution of the haplotypes found in aUzPo via haplotype sharing analyses (Figure 4). These analyses are less impacted by biases due to small population sizes or unidentified maternal relationships in ancient populations, and thus are less prone to artefacts. Although the highest percentages of shared haplotypes for aUzPo were observed in pools of West Siberian Khants/Mansi/Nenets/Selkups (2.8%), South Siberian Altaians/Khakhassians/Shors/Tofalars (2.2%) and Urals populations (Chuvash/Bashkirs, 2.0%), matches were widely distributed across Eurasia. This was consistent with the observation that most haplotypes sequenced in aUzPo were basal and hence, not informative in terms of geographical population affinity. Haplogroup-based analyses suggested that the genetic affinity between aUzPo and present-day West Siberians was partly due to the presence of hg C, implying that the non-basal haplotype C1 found in aUzPo (16189C-16223T-16298C-16325C-16327T, detected in three individuals) could be a clear genetic link with extant Siberian populations. However, the C1 haplotype found in aUz did not belong to hg C1a, the only C1 clade restricted to Asia (characterized by a transition at np 16356 [48]). Indeed, no exact match was found for the C1 haplotype in the comparative database of Eurasian populations (comprising 168,000 haplotypes), although 47 derivatives (showing one to three np differences) were found in extant populations broadly distributed throughout Eurasia (Table S1). Therefore, the C1 haplotype sequenced in aUzPo is currently uninformative about population affinity. In addition, all three aUzPo individuals showed identical C1 haplotypes, which meant that a close maternal kinship between these individuals could not be rejected. Biases due to the overestimation of the hg C1 frequency and small sample size of aUzPo may have led to an overestimation of the genetic affinity with modern-day West Siberians in the hg-based analyses. To account for this, we assumed a scenario of extreme maternal kinship, in which identical haplotypes found in several individuals at the same site (redundant haplotypes) were only counted once (Figure S2A). Under this scenario, the genetic affinity between aUzPo and present-day Western Siberians was less distinctly pronounced (Figure S2B). 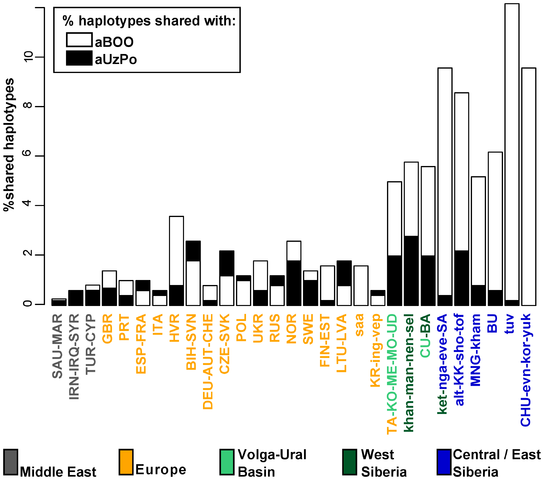 Figure 4. Percentages of haplotypes from aUzPo and aBOO matched in modern-day Eurasian population pools. Comparison of 3,500 uncal. yBP Bol'shoy Oleni Ostrov (aBOO) with extant populations of Eurasia At the 3,500 uncal. yBP site of aBOO, we observed 39% ‘European’ hgs: U5a (26%), U4 (9%), T (4%), and 61% ‘Central/East Siberian’ hgs: C (35%), Z (13%), D (13%). Concordant with this admixed hg make-up, PCA indicated a position close to present-day Siberians (Figure 2). This position did not change when potential maternal relationships among individuals were accounted for by excluding redundant haplotypes (Figure S2B). The genetic relationship between aBOO and Siberians was also evident on the genetic distance map, where the area representing the lowest genetic distance covered a broader area of Siberia than for aUzPo (Figure 3B). The extant populations that showed most genetic similarity to aBOO were found in Central and East Siberia. In contrast, the area of maximum similarity for aUzPo lay in West Siberia (Figure 3A); this observation however could be influenced by low sample size in aUzPo. Haplotype sharing analyses for aBOO confirmed the genetic affinity with modern-day West and Central/East Siberians inferred from the PCA (Figure 4), but also identified a close relationship with the VUB population pool. The distribution of haplotype matches observed in pools of the VUB, West Siberia and Central/East Siberia was partly due to the presence of basal C* (16223T-16298C-16327T) and D* (16223T-16362C) haplotypes in these pools, whereas these types were absent in Middle Eastern and European pools. Central Siberian Tuvinians displayed the highest percentage of shared haplotypes with aBOO (12.2%) although all shared haplotypes belong to hgs C* and D*. A more explicit genetic link between aBOO and extant East Siberians was seen in the presence of the derived C5 haplotype (16148T-16223T-16288C-16298C-16311C-16327T) in aBOO and in one single Buryat individual of Central Siberia [54]. The Z1a haplotype (16129A-16185T-16223T-16224C-16260T-16298C) detected in aBOO had a broad but interesting distribution in Eurasia. It was found in all Central/East Siberian pools except in Tuvinians, but also in the Bashkirs of the Urals, in the VUB pool, as well as in Scandinavian and Baltic populations (Norwegians, Swedes, Finns, Ingrians, Karelians, and the Saami). Although haplotype sharing analyses revealed genetic links between aBOO and extant populations of NEE, a strong genetic differentiation was obvious between aBOO, modern-day NEE and Saami. This genetic discontinuity was further supported by BayeSSC analyses (Figure 5; Table 3). Similarly to aUzPo, a better fit was obtained for the model involving a 10% migration from CE over the last 3,500 years (H1b; ω = 1.00E+0) than for the model of genetic continuity between aBOO and NEE (H0b; ω = 3.86E-10). Comparison among ancient Eurasian populations Previously described populations of hunter-gatherers of Central/East Europe (aHG [12], [14]) and Scandinavia (aPWC, [13]) were characterized by high frequencies and diversity of hg U4, U5a and U5b, which caused the two ancient datasets to group outside the cluster of extant European populations on the PCA plot (Figure 2). This matches previous studies that have shown that genetic continuity between hunter-gatherers and present-day Europeans can be rejected [12]–[13]. Like other European hunter-gatherers, aUzPo is characterized by high frequencies and diversity of hgs U4 and U5, but was genetically differentiated from aHG and aPWC due to the occurrence of hg C. Despite the fact that high frequencies of hgs U5b and V cluster the aHG and aPWC hunter-gatherer groups on the PCA plot (Figure 2), and that these hgs are also common in modern-day Saami, the ‘Saami motif’ is absent from aPWC and genetic continuity between aPWC and modern-day Saami was rejected [13]. Although the aBOO individuals were also characterized by high frequencies of hg U, the group appeared less close to the Palaeolithic/Mesolithic hunter-gatherers aHG and aPWC on the PCA plot than aUzPo. Haplotype sharing analyses (Figure 6) also showed that aBOO shared less haplotypes with aHG and aPWC than aUzPo (4.76% and 0.00%, respectively, versus 9.52% and 36.84%). This observation was confirmed by the analyses of our coalescent simulations, in which a model of genetic continuity between aHG, aPWC and aUzPo (ω = 9.91 E-1; H0d) was better supported than a model of genetic continuity between aHG, aPWC and aBOO (ω = 1.10 E-4; H0e). As demonstrated above, aBOO exhibited greater genetic affinities with extant populations of Siberia than aUzPo. Accordingly, aBOO shared more haplotypes with ancient samples from Siberia aEG (10.87% [55]) and aKUR (7.69% [56]) than aUzPo (0.00% and 7.69%, respectively; Figure 6). DOI: 10.1371/journal.pgen.1003296 |
|
|
|
Post by Admin on Mar 13, 2016 0:55:24 GMT
 The Yiddish language is over one thousand years old and incorporates German, Slavic, and Hebrew elements. The prevalent view claims Yiddish has a German origin, whereas the opposing view posits a Slavic origin with strong Iranian and weak Turkic substrata. One of the major difficulties in deciding between these hypotheses is the unknown geographical origin of Yiddish speaking Ashkenazic Jews (AJs). An analysis of 393 Ashkenazic, Iranian, and mountain Jews and over 600 non-Jewish genomes demonstrated that Greeks, Romans, Iranians, and Turks exhibit the highest genetic similarity with AJs. The Geographic Population Structure (GPS) analysis localized most AJs along major primeval trade routes in northeastern Turkey adjacent to primeval villages with names that may be derived from "Ashkenaz." Iranian and mountain Jews were localized along trade routes on the Turkey's eastern border. Loss of maternal haplogroups was evident in non-Yiddish speaking AJs. Our results suggest that AJs originated from a Slavo-Iranian confederation, which the Jews call "Ashkenazic" (i.e., "Scythian"), though these Jews probably spoke Persian and/or Ossete. This is compatible with linguistic evidence suggesting that Yiddish is a Slavic language created by Irano-Turko-Slavic Jewish merchants along the Silk Roads as a cryptic trade language, spoken only by its originators to gain an advantage in trade. Later, in the 9th century, Yiddish underwent relexification by adopting a new vocabulary that consists of a minority of German and Hebrew and a majority of newly coined Germanoid and Hebroid elements that replaced most of the original Eastern Slavic and Sorbian vocabularies, while keeping the original grammars intact.  There have been other papers which show that Ashkenazi Jews form a separate cluster from gentile whites in the United States. This is important again in the context of biomedical studies attempting to ascertain the genetic roots of particular diseases; population substructure (e.g., Jew vs. non-Jew) may result in confounded associations. Also, one of the authors of the paper is David Goldstein, author of the fascinating Jacob’s Legacy. In any case, on to the PC charts where the real action is. Do note that I’ve resized and added explanatory labels here & there for clarity.  As you can see, there is almost perfect separation between the Jewish and non-Jewish clusters here. PC 1 = first principle component of variation, and PC 2 second principle component of variation. These are the two largest independent dimensions of genetic variance extractable out of the data set. The authors note that there is almost perfect separation along PC 1, and, they note that most of the gentile whites who are closest to the Jews on this PC are of Italian or Eastern Mediterranean origin. This is important later.  As you can see here, the interesting point is that Jewish ancestral quanta is roughly predictive of genetic position. This shouldn’t be that surprising once we know that Jews and non-Jews separate so cleanly (e.g., someone who is biracial would be located between their two parent racial clusters on any plot), but it is striking nonetheless in reaffirming the genetic reality of Jewishness. The United States has an easy genealogical history for Jews, as the ancestors of all self-identified American Jews today arrived on the order of 4-6 generations ago. These individuals were likely Jewish relatively far back in their lineages, and admixture is easy to recall because it is of recent vintage. In conclusion, we show that, at least in the context of the studied sample, it is possible to predict full Ashkenazi Jewish ancestry with 100% sensitivity and 100% specificity, although it should be noted that the exact dividing line between a Jewish and non-Jewish cluster will vary across sample sets which in practice would reduce the accuracy of the prediction. While the full historical demographic explanations for this distinction remain to be resolved, it is clear that the genomes of individuals with full Ashkenazi Jewish ancestry carry an unambiguous signature of their Jewish heritage, and this seems more likely to be due to their specific Middle Eastern ancestry than to inbreeding. |
|
|
|
Post by Admin on Mar 19, 2016 0:54:52 GMT
 The genome map was drawn using MDS with an RH-free dataset (Supporting Methods and Discussion S1). The genome map in Fig. 2A showed that the Yoruban, Caucasian, and Amerindian populations lined up first, and then Northern and Southern Asian populations were merged in East Asia in Fig. 2B. In the extended genome map for Northern and Southern Asia, the merged pattern became clearer than that of the whole populations where the MH (Mongolian) population lined up to JPT (Tokyo) and JJ (JeJu) populations, while the CB (Cambodian) population lined up to the VN (Vietnamese), VK (Vietnamese-Korean), CHB (Peking), and JL (Jilin) populations. 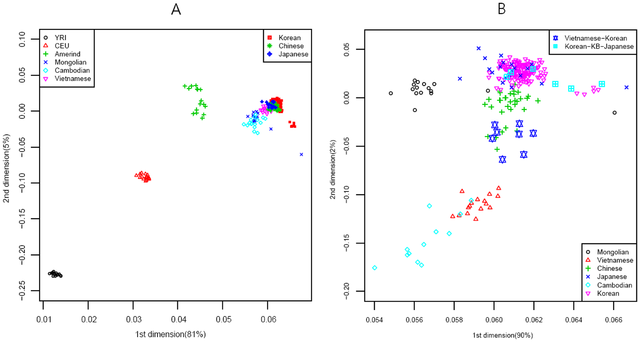 Figure 2. Genome Map of World (A) and Asia (B). doi:10.1371/journal.pone.0011855.g002 In our analysis of genetics structure by STRUCTURE program [16], four populations displayed perfect population membership probabilities, the Africans, Caucasians, Amerindians, and Northern Asians, and three populations displayed partial membership, the Mongolians, Vietnamese, and Cambodians (Fig. 3). Substantial admixture patterns between populations were also observed for Mongolians and Caucasians, and Amerindians with East Asians and Caucasians. 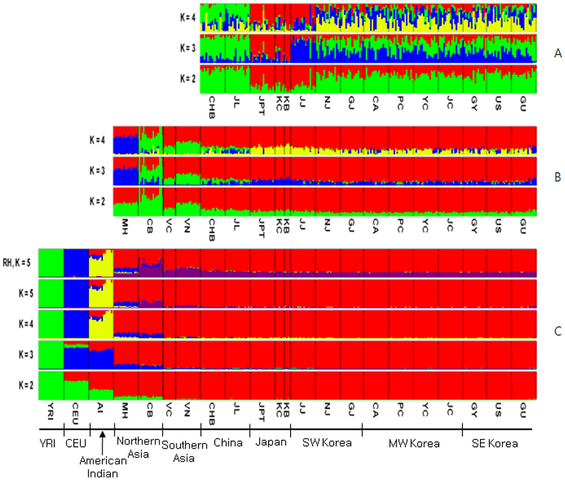 Figure 3. Genetic Structure. A)World, B)Asia, C)Korea, Japan and China. Note that the plot with a recombination hotspot (RH) of K = 5 on the left is shown to compare with the same plot without the RH effect on the right where the purple bands were spread over all Asian populations. doi:10.1371/journal.pone.0011855.g003 Genome map and an NJ tree [18], [19] for Korea-China-Japan are shown in Fig. 4, where the genome map is a further extension of the World and Asian map (Fig. 2). In the figure, dots representing the Chinese are located at the bottom and those for Japanese are at the top, while some outliers in SE Korea and Kobe in Japan are shown on the right hand side. Of the ten historical regions in South Korea, some in SW Korea overlap with those of Japan, while most of the MW Korean regions are located at the center of the genome map. Similarly, in the NJ tree, nodes for SW Korea are close to those in Japan, MW Korea is close to China, and SE Korea is located at the right hand side of the tree. 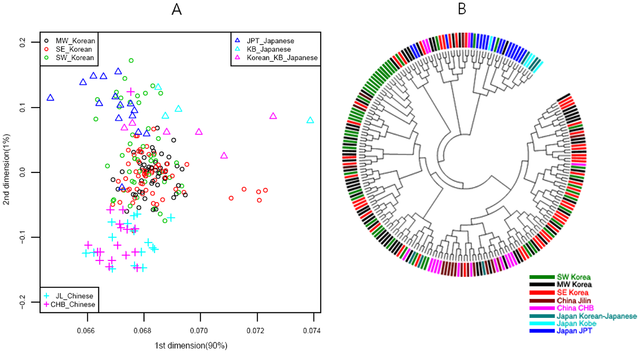 Figure 4. MDS and NJ Tree of Korean, Chinese and Japanese. A)MDS(multi-dimensional scaling) [17], B)NJ Tree. In the NJ tree, samples for Japan, Kobe-Japan, SW Korea, SE Korea, MW Korea, China, Jinlin, and Korean-Japanese are blue, cyan, green, purple, black, brown and dark green, respectively. doi:10.1371/journal.pone.0011855.g004 The gene flow events of the three selected models for SW, MW and SE Korea can be assessed using the genome map. The populations in Model I (SW Korea) are closer to Mongolians than are the other two regions in the genome map (Fig. 2B). Historically, some of the loyal families and their subjects in the Goguryeo Empire moved to this region and formed the BaekJae Empire in BC18-22. This region also showed connections with populations in Tokyo (JPT), as illustrated in Fig. 4. Certain outliers in Model II (SE Korea) display some similarity to the people of Kobe, a port city near Osaka, indicating that there may have been links between the two regions. In addition, considering that the SE Korea region has some connections with Siberian lineages, with respect to grave patterns and culture, it is possible that the outliers in the GU and Kobe (KB) populations could be of Siberian lineage. On the other hand, the GR and US populations showed average signals in the Korean Peninsula. Historically, the Kaya Empire, with its southern lineages, was formed in the GR region and then the Shilla and Kaya Empires became united around AD532. Very recently, the US region became one of the rapidly developing regions, and people from other provinces moved to this region. This might explain why it shows an average signal in South Korea. Model III (MW Korea): the Middle West area formed a melting pot in the Korean Peninsula because populations moving from South to North, North to South, and from Eastern China, including the SanDung peninsula, to the Middle West in Korea all came together in this region. In the genome map, the signals for MW Korea are also close to those for Peking (CHB) in China. The overall result for the Korea-Japan-China genome map indicates that some signals for Mongolia and Siberia remain in SW Korea and SE Korea, respectively, while MW Korea displays an average signal for South Korea. Current populations in the Korean Peninsula were formed through interactions between populations from Southern and Northern Asia. Therefore, the goal of this study was to determine whether there are still genetic signals remaining in the Korean Peninsula using autosomal genome-wide SNPs, which are 100 times more frequent than those in lesser recombining DNA, such as mtDNA, X and Y chromosomes. Interestingly, some of the historical regions and the regionally separated island, Jeju (JJ) and Gyeongju (GU) in Korea still harbor some old signals from Northern populations, while few signals remain from Southern populations. In this study, we reconstructed the genome map using data from 320 individuals in 24 groups after correcting and re-evaluating all bias parameters (Supporting Methods and Discussion S1, Fig. S1, S2, S3 and S4). Interestingly, the genome map was similar to the physical map of Europe and Asia, a relationship that was reported previously in a correlation experiment between physical distances and genetic variations by Mountain and Cavalli-Sforza [12]. In addition, genetic structure analysis using the STRUCTURE method revealed the existence of five major populations, African, Caucasian, Amerindian, North-East Asian, and Southern Asian. Therefore, in between, there were significant admixtures such as Mongolian with Caucasian, Vietnamese (or Cambodian) with unknown Southern original settlers, and Amerindians with both North-East Asians and Caucasians (Fig. 2). Thus, the genome map could be used to estimate the relative evolution times between populations and genome interactions (or gene flows) (Fig. 4) such as Korean-Vietnamese, between Vietnam and Korea; Korean-Japanese, between Korea and Japan; Mongolian between Amerindians and North-East Asians; Chinese between Koreans and Vietnamese; and Vietnamese between Cambodians and Chinese. In particular, through both the genome map and STRUCTURE results, the evolutionary relationship between Koreans, Japanese, and Chinese has become clearer since it can be seen that the North-East Chinese (JL) and the Peking Chinese (CHB) are relatively closely related, and the Japanese (JPT) and Korea-JeJu (JJ) are also fairly close (Fig. 2). Jung J, Kang H, Cho YS, Oh JH, Ryu MH, Chung HW, et al. (2010) Gene Flow between the Korean Peninsula and Its Neighboring Countries. PLoS ONE 5(7): e11855. doi:10.1371/journal.pone.0011855 |
|

