|
|
Post by Admin on Dec 4, 2019 18:23:37 GMT
Different Immune Responses in African and Eurasian Populations The migration of our human ancestors out of Africa implied the exposure to different types of infectious diseases (Box 2 and Figure 3). One study tested the responsiveness of human macrophages to pathogenic bacteria in vitro, finding that almost 10% of the genes present in human macrophages infected with the bacteria Salmonella typhimurium or L. monocytogenes present different regulatory responses directly linked to the lineage of the donors and, also, that macrophages obtained from individuals of African origin display enhanced bactericidal activity compared with those from individuals of European lineage [32]. The trend towards lower inflammatory responses in European populations is strengthened by the fixation of a TLR1 gene variant that results in lower proinflammatory gene expression in populations with a European ancestry compared with those with an African one [33]. The largest population differences in gene expression between Africans and Europeans have been found in the macrophage receptor with collagenous structure (MARCO), a protein implicated in responses against viral infections and TLR-induced dendritic cell activation [34], the chemokine receptor CX3CR1, which mediates effector lymphocyte functions, and also several IFN-stimulated genes [35]. West Eurasian populations present a high frequency of TIRAP Ser180Leu SNPs [36]. TIRAP is an adaptor protein in TLR2 and TLR4 signaling pathways, involved in inflammatory responses and cytokine production. The heterozygous expression of the Ser180Leu SNP is protective against invasive pneumococcal disease, bacteremia, malaria, and tuberculosis, as shown in a case–control study of 6106 individuals from the UK, Vietnam, and several African countries, and it is associated with lower TLR2 signaling in humans [36]. This variant is considered to be a consequence of the natural selection that may have taken place in an early period following the migration of modern humans out of Africa [37]. Box 2  Figure 3 Map of Human Migrations. European populations present a selective adaptation of the IFN gene that allows a high production of IFN-γ in infectious scenarios due to the positive selection of IFNG variants +5173G and +874T; this suggests the existence of strong environmental pressures linked to higher IFN-γ concentrations in plasma during MTB infection in European individuals relative to other populations [38,39]. In line with this, a database meta-analysis showed that individuals expressing the +874T/A variant of IFNG presented higher susceptibility to tuberculosis MTB infection than individuals without it, which might be considered a putative prognostic factor for the development of tuberculosis [40], although this remains to be robustly demonstrated. When humans ventured out of Africa, they faced different types of pathogens than the communities that stayed in the African continent. With time, the series of events faced by diverse populations has generated differences in the immune responses to pathogens in the populations with an African or Eurasian origin and which have spread throughout the world. Colonization of New World and Immunity in the American Continent The indigenous populations of South America are descendants of migrating populations of North-East Asians that crossed the Bering Strait around 20 000 years ago [41]. Five centuries ago, European settlers disembarked on the American continent, bringing a large collection of pathogens such as those causing measles, pneumonic plague, and influenza infections, which the indigenous populations had never faced before. These diseases rapidly spread and caused mortality rates above 90% [42]. The consequences of these pandemics are still visible in current populations; one report studied DNA from the bones of 25 ancient humans from the Tsimshian community, living in the British Columbia region in Canada until the 15th century, identifying marks of positive selection in a number of immune-related variants [43]. Specifically, the HLA-DQA1 variant was present in almost the totality of Tsimshian individuals, but only in one third of present-day humans studied; this suggested that ancient American genomes were evolutionarily selected to respond to local diseases but not to fight against pathogens brought by the Europeans [43]. Another study compiled information on infectious diseases that have killed more than 10 000 individuals among 59 indigenous communities of the Amazonia in the past two centuries, showing that the mortality rates and the incidence of infectious diseases rapidly decayed within the time following the first encounter with the pathogen, compatible with genetic adaptation [44]. European colonizers underwent purifying selection in situations of intense pressure. Such scenarios were documented when Dutch colonists migrated to Surinam and encountered epidemics of yellow and typhoid fever that caused a 60% mortality rate among settlers [45]. Variations in the frequencies of C3, GLO, and HLA-B genes among the descendants were not likely caused by genetic drift, but rather, it has been proposed that these populations were probably selected through genetic control of survival to the epidemics [45] (Box 3). Box 3 Africans and Americans with an African ancestry present a much higher number of genetic variants related to robust inflammatory reactions, increased cytokine secretion, and bactericidal activities compared with the other populations [32], including more than 250 genes with traces of recent selection, such as IL1A and IL1B gene variants [46]. The degree of African ancestry, analyzed by fine-mapping analysis refined to the Duffy-null allele of rs2814778, was correlated with an increased amount of the proinflammatory chemokines CCL2 and CCL11 in plasma relative to controls [12]. A study involving 12 000 African American and Hispanic American women found that the higher values of C-reactive protein (CRP) in blood found in these populations compared with European Americans were related to a CRP-associated variant of triggering receptors expressed by myeloid cells 2 (TREM2) [47]. Moreover, comparison of health record data from individuals with connective tissue diseases, including rheumatoid arthritis and systemic lupus erythematosus (SLE), as well as atherosclerotic cardiovascular disease from almost 300 000 African American and European American adults was conducted; the study reported for the latter, a prevalence of atherosclerotic cardiovascular disease in 29.7% African Americans (particularly high in young individuals), relative to 14.7% in European Americans [48]. These studies highlight certain genetic links to inflammatory predisposition/manifestation. However, increased proinflammatory activity is a double-edged sword. In the absence of regular pathogen challenges that require maintained modulation of the balance between inflammation and suppression of the immune response, the organism can overreact to inflammatory stimuli and trigger exacerbated responses. For instance, descendants of African populations generally present higher susceptibility to a variety of autoimmune syndromes such as inflammation-associated carcinomas, lupus, asthma, and multiple sclerosis (MS), the overall prevalence of which is up to three times higher in individuals with African ancestry relative to individuals with European ancestry [31,49,50]. There are extensive differences in immune cell gene expression between Americans with African and European ancestry. The increased proinflammatory responses observed in American individuals relative to other populations might be beneficial to combatting infections, but might also increase the chances of developing inflammatory and autoimmune disorders, which warrants further investigation. |
|
|
|
Post by Admin on Dec 5, 2019 17:27:59 GMT
Nonpathogenic Microbes: Mutualistic Bacteria and Viruses Our gastrointestinal tract provides residence to both beneficial and potentially pathogenic microorganisms, harboring ten million different microbial genes in the human fecal microbiome [51]. The microbiome has its own evolutionary scenario across different populations with divergent lifestyles, nutrition, and exposure to environmental agents, generating extraordinary heterogeneity. The ongoing process of ‘lifestyle Westernization’ of different societies has an important impact on the mutualistic relationships between humans and commensal organisms worldwide. African tribes are adopting Western subsistence patterns, leading to remarkable changes in the composition of their microbiota [52]. The comparison of the intestinal flora of the BaAka hunter-gatherers and the Bantu agriculturalists (both from the Central Africa Republic), with a group of US-born African Americans showed a great example of the evolution of the human microbiome [52]. Specifically, the Bantu, still engaged in hunting, have a greater bacterial gut diversity than their BaAka neighbors, who left the jungle for agriculture, and even more than urbanized westerners (US African-Americans) [52]. This reduced microbiota diversity in Western societies has been associated with a higher incidence of the so-called ‘diseases of civilization’ such as cardiovascular diseases, diabetes, obesity, and autoimmune disorders, which are very unusual in hunter-gatherer societies compared with communities living a Western-type lifestyle [52,53]. Although viruses are mainly seen as pathogenic agents, they also play a fundamental role in the evolution and maturation of the human immune system [54,55]. Approximately 8% of the human genome is composed of endogenous retroviruses (ERVs), sequences derived from past retroviral infections and permanently inserted into different regions of the human genome [56]. One study showed that ERVs played a central role in the induction of IFN-dependent immune responses and that the removal of one or more of these viral DNA elements in the HeLa human cell line severely impaired the recruitment of transcription factors necessary to trigger the expression of IFNG against vaccinia virus infection relative to controls [57]. Viruses can also influence the severity of infections caused by other viruses. For example, cytomegalovirus infection in HIV-1 seropositive humans can potentiate the effects of HIV-1 infection by expanding the pool of circulating regulatory T cells (immunosuppressive); these were shown to inhibit the proliferation of autologous peripheral blood mononuclear cell (PBMC) in response to cytomegalovirus infection in vitro [58]. In one study, patients with chronic hepatitis C virus (HCV) infection and hepatitis A virus (HAV) superinfection presented lower titers of HCV RNA than patients harboring only HCV, suggesting that HCV replication might be potentially suppressed during HAV infection [59], although this will still require further investigation. The relationships between humans and pathogenic or nonpathogenic organisms are extraordinarily complex and include tripartite evolutionary interactions between humans and microbes competing with each other. This is the case of parasites that infect other parasites, such as bacteriophage viruses, that can influence the outcome of bacterial infections. For example, in a cohort of individuals with chronic wounds, a report showed that the phage Pf, which coexists with Pseudomonas aeruginosa in infected wounds, triggered the production of type I IFN, the inhibition of tumor necrosis factor (TNF) production, and the suppression of phagocytosis in human primary monocytes and mouse bone marrow-derived macrophages, dampening the antibacterial response and promoting the bacterial infection [60]. However, bacteriophages can also provide protection to the human host by directly attacking pathogenic bacteria and by upregulating in human PBMCs the expression of proinflammatory genes such as IL1A, IL1B, IL6, TNFA, CXCL1, and CXCL5, as shown for several S. aureus and P. aeruginosa phages, including PNM, LUZ19, 14-1, and GE-vB_Pae-Kakheti25 [61]. Cooperative relationships between organisms are evolutionary processes themselves. The way microorganisms and their hosts associate can lead to interactions of mutualism, in which the interplay may be so intimate as to provide benefit for each party and influence immune responses against different types of pathogens. A Role for Ancestry in the Susceptibility to Autoimmune Diseases A great number of humans live far away from the original settlements of their ancestors and are subject to radically different environmental conditions. Between two and three million people with European genealogy suffer from autoimmune diseases, the prevalence of which is also increasing in other populations across the globe [62]. There is rising evidence that the emergence of autoimmune diseases is associated with the presence of a number of immune-related alleles that have been selected via evolutionary processes; and, furthermore, that the contrasting differences in the prevalence of autoimmune diseases between populations may be a result of different selective pressures [63]. Alleles associated with inflammatory diseases that present marks of modern positive selection include the risk allele FUT2 at rs601338 for Crohn’s disease (CD) or the risk variant SH2B3 at rs3184504 for celiac disease [64]; such variants have been linked to the development of several human autoimmune diseases, such as type 1 diabetes, MS, and celiac disease [64,65]. The analysis of the integrated haplotype score [66] of loci associated with SLE that might provide protection against infections, such as TNIP1, ITGAM, PTPN22, TNFSF4, UHRF1BP1, TET3-DGUOK, and BLK, has suggested that these loci exhibit robust signs of positive selection [67] (Figure 4, Key Figure and Table 1). African and Asian human populations exposed to Trypanosoma brucei or Plasmodium sp. have presented positive selection of SNPs in the APOL1 and FCGR2B genes; indeed, by enhancing human macrophage-mediated phagocytosis of infected erythrocytes, despite their association with an SLE predisposition, these SNPs have been associated with protective roles against sleeping sickness and malaria, respectively [68,69].  Figure 4 Key Figure. Impact of Ancestry on Immunity An analysis of human loci associated with inflammatory bowel disease (IBD) concluded that the majority of the loci associated with CD are also linked with a higher risk of developing ulcerative colitis [70] (Table 1). Many of these loci were also associated with the development of other autoimmune diseases, namely psoriasis and ankylosing spondylitis [62]. PBMCs from individuals carrying the SH2B3 variant rs3184504*A (associated with a risk for developing CD) [71] presented higher production of IL-6 and IL-1β after stimulation with lipopolysaccharide or muramyl dipeptide due to enhanced activity of the NOD2 inflammasome pathway relative to controls [72]; this has suggested an immune-related role for SH2B3 in the context of bacterial infections, which might help explain the positive selection of SH2B3 approximately 1500 years ago [72]. Others found an association between genetic variants in NOD2, CD14, and increased susceptibility to CD [73], in line with previous results showing that mutations in NOD2 and TLR4/CD14 are related to an increased risk of developing IBD [74]. From another angle, changes in hygiene patterns seen in the past two centuries brought vast improvements in sanitation, drinking water, and garbage collection, which greatly reduced the exposure to many infectious diseases. However, these conditions may have reduced the exposure to viral and microbial agents that help the immune system to develop tolerance during childhood. The hygiene hypothesis proposes that the lack of exposure to microbial agents in the early stages of life is related to a higher risk of developing hypersensitivity reactions, based on the fact that children that are exposed to higher amounts of microbial stimuli (e.g., by growing on farms) are less prone to develop allergies and asthma [75,76]. Moreover, reduced exposure to infectious agents can have a much wider effect than initially believed. Lack of exposure to microbes in childhood can cause aberrant responses to infection and potentiate the effects of ETV6–RUNX1 mutations in the pathogenesis of acute lymphoblastic leukemia [77]. By contrast, a meta-analysis of six observational studies, including 1902 participants, showed a correlation between low Helicobacter pylori infection and MS, suggesting that low H. pylori prevalence might be a putative protective factor in MS, although this remains to be experimentally validated [78]. One study also reported that antibodies against Toxoplasma gondii were detected less often in patients with MS compared with healthy controls [79]. However, these findings warrant further and robust investigation. Overall, it is clear that evolutionary processes can drive the fixation of genetic variations that increase (or decrease) our defense against infections upon sensing microbial ligands, but can also lead to a greater risk of developing certain autoimmune diseases in which endogenous ligands can cause tissue damage and inflammation. The Potential Role of Epigenetic Inheritance A growing number of reports suggest that inheritance is not always governed by classical Darwinian evolutionary processes. Exposure to certain environmental stimuli can cause effects in the progeny of an exposed individual, even though the stimuli are no longer present. This type of transgenerational inheritance might be explained through the effects of epigenetic processes, which are hypothesized to be transmitted through the germline and passed on to the offspring [80]. For example, the worm Caenorhabditis elegans can transmit improved resistance to infections to pathogenic bacteria to their offspring through alterations of the histone landscape [81]. Indian meal moths exposed to low doses of virus are subsequently less susceptible to viral challenge, a protection offered to their offspring as well [82]. Transgenerational inheritance of diverse traits has also been observed in mice, in which the variation of the color of the coat is passed on the next generation [83,84]. Offspring of male rats subjected to a high-fat diet present glucose intolerance and reduced insulin secretion, linked to reduced methylation at the Il13ra2 gene relative to controls [85]; and mice fed scorpions are more resistant to a challenge with scorpion venom than mice on a normal diet [86]. Since infections are one of the strongest factors impacting survival, it is conceivable that transgenerational transmission of traits in mammals, including humans, evolved to improve host defense. The number of studies of the potential role of epigenetic inheritance in shaping the human immune system is still scarce. However, different experiences undergone by certain communities indicate that these mechanisms might be important. For example, the babies of pregnant women who suffered during the early stages of pregnancy (the effects of the Dutch Hunger Winter in 1944), 60 years later showed reduced DNA methylation marks in several genes that control metabolism and cell differentiation during development, such as IGF2, PIM3, TXNIP, ABCG1, PFKFB3, and METTL8, compared with their siblings [87,88]. This was related with higher rates of obesity, heart disease, cancer, and depression in individuals whose pregnant mothers suffered the effects of the famine [87,88]. Some of these effects seemed to be present in the progeny of this group, that is, in the grandchildren of those who had passed the famine during pregnancy [89]. The rapid growth in the number of reports covering the impact of epigenetic mechanisms in different human processes warrants further and robust studies on the impact of epigenetic inheritance in shaping the evolution of the human immune system. Trends in Immunology | VOLUME 40, ISSUE 12, P1105-1119, DECEMBER 01, 2019 |
|
|
|
Post by Admin on Dec 6, 2019 17:37:43 GMT
 Researchers have teased out evidence suggesting the ancestors of modern-day sub-Saharan Africans interbred with an archaic human group. At the same time, though, they found no evidence of non-Neanderthal or non-Denisovan archaic admixture among Andaman Islanders, which had been previously suggested. As anatomically modern humans migrated from Africa, they came into contact and interbred with archaic human groups. Non-Africans are estimated to have inherited about 2 percent of their DNA from Neanderthals, while Melanesian and aboriginal Australians have inherited about 4 percent to 5 percent of their DNA from Denisovans, and yet other populations, like the Andaman Islanders, were thought to have interbred with as-yet-unknown hominins. But as archaic humans also lived in sub-Saharan Africa around the same time and in the same places as a modern humans, there may have been opportunities for admixture inside Africa, as well. To study this, Jeffrey Wall, a researcher at the University of California, San Francisco and his colleagues used a dataset of genomes from more than 1,600 individuals from across the globe, recently reported by the GenomeAsia 100K Consortium, to search for signs of archaic admixture. As they reported today in the American Journal of Human Genetics, the researchers found evidence of such admixture among sub-Saharan African individuals based on a linkage disequilibrium approach. They did not find, though, evidence for interbreeding with archaic humans among Andaman Islanders or people from the island of Flores. "We note that the extent of admixture between modern and archaic humans, while clearly of historical and evolutionary interest, also has direct implications for human disease genetics," Well and his colleagues wrote in their paper.  The researchers searched for signs of admixture with archaic human groups without relying on genome sequence data from those groups. Instead, they used a linkage disequilibrium approach and searched for long, diverged haplotypes within the 1,667 genomes from the GenomeAsia 100K Project (GAsP) pilot dataset, which includes not only individuals from Asia but also from Africa, Europe, and the Middle East. The researchers considered the long, diverged haplotypes they found as candidate regions for archaic human introgression and dubbed them "putative ghost haplotypes," or PGHs. After validating their approach by searching for known Denisovan-derived tracts within non-African samples from GAsP, the researchers then applied it to their full GAsP cohort. Through this, they identified 2,319 autosomal PGHs.  The average number of PGHs, they noted, was much higher — between five and 15 times — among sub-Saharan Africans than non-Africans. It was also higher than the researchers had expected based on simulated datasets. This suggested that a major archaic admixture event may have taken place in sub-Saharan Africa. Based on differences in PGHs among genomes from Khoesans, Central African Pygmies, and West, East, and North Africans, they further said that this admixture event likely took place in the ancestors of modern-day Khoesans before spreading to other modern human populations. The researchers cautioned, though, that their approach cannot fully disentangle models of long-term isolation and population structure from their proposed archaic admixture scenario, and said that additional sequencing and analysis of genomes from Central and Southern African groups will be needed. Meanwhile, they found few signs of non-Neanderthal, non-Denisovan archaic admixture among non-Africans, including South Asians and Melanesians. A previous study had reported a signal of unknown archaic admixture among Andaman Islanders, but the UCSF-led team could not detect any, despite deep sampling in the region. Additionally, they found no signs of additional archaic admixture among individuals from Flores, where the ancient hominin Homo floresiensis was uncovered. Recent work has demonstrated that two archaic human groups (Neanderthals and Denisovans) interbred with modern humans and contributed to the contemporary human gene pool. These findings relied on the availability of high-coverage genomes from both Neanderthals and Denisovans. Here we search for evidence of archaic admixture from a worldwide panel of 1,667 individuals using an approach that does not require the presence of an archaic human reference genome. We find no evidence for archaic admixture in the Andaman Islands, as previously claimed, or on the island of Flores, where Homo floresiensis fossils have been found. However, we do find evidence for at least one archaic admixture event in sub-Saharan Africa, with the strongest signal in Khoesan and Pygmy individuals from Southern and Central Africa. The locations of these putative archaic admixture tracts are weighted against functional regions of the genome, consistent with the long-term effects of purifying selection against introgressed genetic material. AJHG VOLUME 105, ISSUE 6, P1254-1261, DECEMBER 05, 2019 |
|
|
|
Post by Admin on Dec 24, 2019 21:57:56 GMT
A new study from the lab of Harvard researcher Yakeel Quiroz, PhD, has suggested a new target for drugs that might have the potential to slow down or even stop Alzheimer’s disease in its tracks. 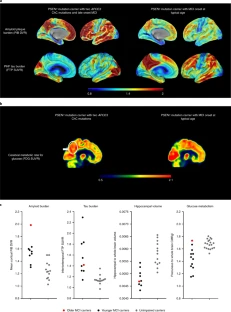 Fig. 1: Brain imaging shows limited tau pathology and neurodegeneration despite high amyoid-β plaque burden in an individual homozygous for APOE3ch. A family with early-onset disease — and one exception Dr. Quiroz, her longtime colleague Dr. Francisco Lopera, and first author Dr. Joseph Arboleda-Velasquez have been studying a large family in Colombia, South America, some of whom have a mutation in the presenilin 1 gene that causes early-onset Alzheimer’s disease. Over 1,000 people in this family are affected by the mutation. Among these family members, early symptoms of Alzheimer’s, such as memory loss and word-finding difficulties, almost always develop around age 44, and dementia follows at around age 49. Sometimes individuals may develop these symptoms or dementia one, two, or even three years later. But not 10 or 20 years later — and certainly not 30 years later. Yet one individual — a woman in her 70s with this genetic mutation — is only now starting to show symptoms. The study, reported in the November 2019 issue of Nature Medicine, is a case report and extensive analysis of this one woman. The APOE gene can modify your risk of Alzheimer’s Many people have read or heard about variations in the APOE gene as a risk factor for Alzheimer’s. Interestingly, in their inquiry into why this woman with a mutation for early-onset Alzheimer’s had not yet developed dementia, the researchers found that she had an additional mutation in her APOE gene. 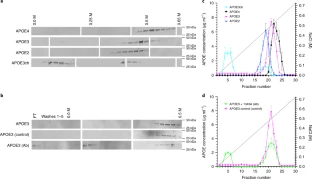 Fig. 2: The Christchurch mutation impairs the heparin binding of APOE. APOE has been linked to ordinary, late-onset Alzheimer’s disease and comes in three common forms. Most people, about 70% to 75%, have APOE3. About 15% to 20% of people have an APOE4 gene, and about 5% to 10% of people have an APOE2 gene. If you have one APOE4 gene, your risk of developing Alzheimer’s disease is three to four times more likely than if you only have APOE3 genes. If you have one APOE2 gene, your risk of developing Alzheimer’s disease is somewhat less than if you only have APOE3 genes. This woman’s mutation of her APOE gene is an unusual variant called APOE3Christchurch (APOE3ch), named after the New Zealand city where it was first discovered. Even more unusual is the fact that she had two versions of this mutation, meaning that both her father and her mother gave it to her. The researchers wondered if this APOE3ch mutation could be the cause of her resistance to Alzheimer’s disease. 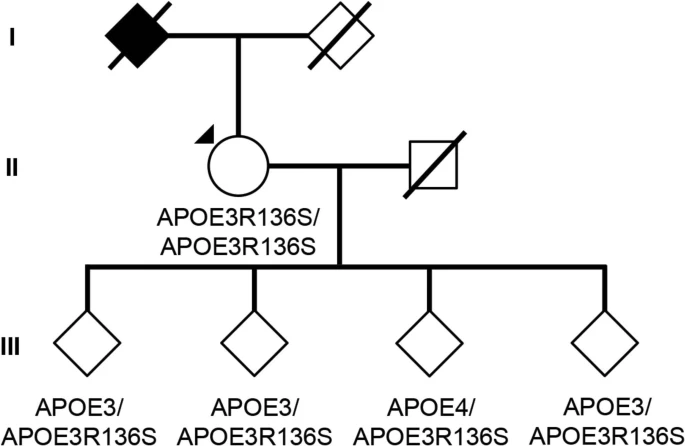 Extended Data Fig. 1 Subject’s genealogy. Resistance to tau Another piece of the puzzle relates to an abnormal protein called tau. Tau is associated with the destruction of brain cells in Alzheimer’s disease. Tau is thought to accumulate in the brain after amyloid protein — the pathologic hallmark of Alzheimer’s disease — forms plaques. Although her brain was full of abnormal amyloid plaques — even more so than most people with full-blown Alzheimer’s dementia — she had relatively little tau. Now the question was, could the APOE3ch mutation be related to the small amounts of tau protein? Although the answer is far from settled, the researchers did uncover some clues through laboratory experiments. Their findings suggest that the APOE3ch mutation may reduce the uptake of tau in brain cells. In addition, they were able to produce similar beneficial results using a special protein they created in the laboratory to try to mimic the effects of the APOE3ch mutation. 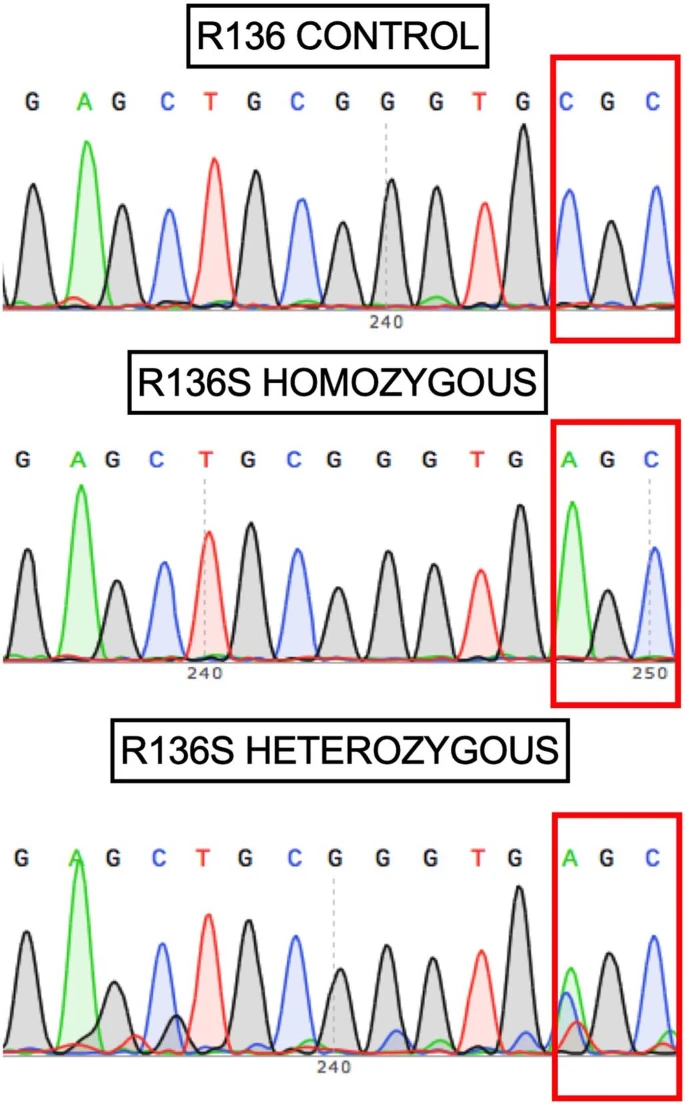 Extended Data Fig. 2 Sanger DNA sequencing of homozygous APOEch carrier individual. Where we are now In brief, these Harvard researchers have a viable hypothesis to explain why this woman has been highly resistant to developing Alzheimer’s disease dementia. Moreover, their work suggests a possible path to a treatment that could be beneficial for all forms of Alzheimer’s disease. We are still years away from a human treatment. The next step will be to try to treat laboratory models of Alzheimer’s disease in rodents, and then clinical trials in people with the disease after that. But in my view, this paper has provided the scientific community with a clue that may lead us to an eventual cure for Alzheimer’s disease.  Extended Data Fig. 3 APOE3ch modulates Aβ aggregation. Abstract We identified a PSEN1 (presenilin 1) mutation carrier from the world’s largest autosomal dominant Alzheimer’s disease kindred, who did not develop mild cognitive impairment until her seventies, three decades after the expected age of clinical onset. The individual had two copies of the APOE3 Christchurch (R136S) mutation, unusually high brain amyloid levels and limited tau and neurodegenerative measurements. Our findings have implications for the role of APOE in the pathogenesis, treatment and prevention of Alzheimer’s disease. Nature Medicine volume 25, pages1680–1683(2019) |
|
|
|
Post by Admin on Dec 26, 2019 20:03:25 GMT
As our primary interface with the environment, the immune system is thought to have evolved under strong selective pressure from pathogens (Barreiro and Quintana-Murci, 2010, Fumagalli et al., 2011, Karlsson et al., 2014). When human populations migrated out of Africa, they encountered markedly different pathogenic environments, likely resulting in population-specific selection on the immune response (Barreiro and Quintana-Murci, 2010, Fumagalli et al., 2011, Karlsson et al., 2014). Substantial evidence supports this hypothesis at the genetic level. However, we still know little about the extent to which neutral or adaptive inter-population genetic differences affect the actual immune response to pathogens. Addressing this gap is not only important for understanding recent human evolution, but may also help reveal the molecular basis of ancestry-related differences in disease susceptibility. Individuals from different populations vary considerably in their susceptibility to many infectious diseases, chronic inflammatory disorders, and autoimmune disorders. For tuberculosis, systemic lupus erythematosus, systemic sclerosis, psoriasis, and septicemia, African American (AA) and European American (EA) individuals exhibit an up to 3-fold difference in prevalence (reviewed in Brinkworth and Barreiro, 2014, Pennington et al., 2009, Richardus and Kunst, 2001). These observations argue in favor of significant ancestry-related differences in immune response, especially in susceptibility to inflammation (Pennington et al., 2009, Richardus and Kunst, 2001).  Figure 1 European and African Ancestry-Associated Differences in Immune Response Such differences almost certainly involve major contributions from the environment. However, genome-wide association studies (GWAS) also support a key role for genetic factors, as many of the GWAS-variants associated with infectious, autoimmune, and inflammatory diseases present extreme differences in allele frequency (Fst > 0.4) between human populations, again supporting a possible history of population-specific selection (Brinkworth and Barreiro, 2014). GWAS results also indicate that susceptibility to many common immune-related diseases is primarily controlled by non-coding variants (Gusev et al., 2014, Hindorff et al., 2009, Schaub et al., 2012). Thus, many ancestry-related differences in disease susceptibility may result from genetically controlled transcriptional differences in immune responses to inflammatory signals. This idea is consistent with recent expression quantitative trait locus (eQTL) mapping studies in innate immune cells exposed to immune antigens or live infectious agents (Barreiro et al., 2012, Çalışkan et al., 2015, Fairfax et al., 2014, Lee et al., 2014). Such immune “response eQTL” studies have identified hundreds of genetic variants that both explain variation in the host immune response and are significantly enriched among GWAS-associated loci. However, because studies to date have mostly focused on individuals of European ancestry, the degree to which such variants contribute to population differences in the immune response remains unclear. Here, we report an RNA-sequencing (RNA-seq)-based immune response eQTL study to test for the effects of African versus European ancestry on the transcriptional response to several live bacterial pathogens. We integrate statistical and evolutionary genetic analyses with primary macrophage gene expression levels, before and after infection, to characterize ancestry-related differences in the immune response. Our analyses address three fundamental questions about recent evolution in the human immune system: (1) the degree to which innate immune responses are differentiated by European versus African ancestry, (2) the genetic variants that account for such differences, and (3) the evolutionary mechanisms (neutral genetic drift versus positive selection) that led to their establishment in modern human populations. |
|

