|
|
Post by Admin on Jan 9, 2020 18:22:13 GMT
In a new paper this week, doctors at the Mayo Clinic say they’ve uncovered the cause of a mysterious heart condition that had suddenly killed over a dozen young, healthy members of a tight-knit Amish community. The culprit? A previously undiscovered genetic mutation that runs in families. Study author Michael Ackerman, a cardiologist and professor at the Mayo Clinic College of Medicine and Science, is also director of the Clinic’s Sudden Death Genomics Laboratory. For years, the lab has investigated cases in which seemingly healthy people died with no clear cause, hoping to unearth new ways our genes can send us to an early grave. In many of these cases, people’s hearts simply stopped beating, a condition otherwise known as cardiac arrest.  “The medical examiner first contacted me and my research team over 15 years ago, after the deaths of two Amish siblings during recreational play over four months’ time,” Ackerman told Gizmodo via email. “For me, in situations like these, it is either foul play or genetic. [But] there was no way based on interviews with the family that this was foul play, so we searched for the genetic cause of sudden cardiac death.” As is often the case with smaller, isolated communities of people, the Amish have more in common genetically with one another than people living in a typical modern community do with their neighbors. Unfortunately, the less genetically diverse a population is, the easier it is for harmful genetic conditions to emerge and be passed down to the next generation. These conditions are often recessive, meaning it takes having two copies of the unlucky genetic variation—one inherited from each parent—for symptoms to show up. Those who carry just one copy of the bad variation usually end up with no health problems, and even if they have children with another carrier, there’s only a 25 percent chance a child of theirs will have both copies. From the start, Ackerman and his team suspected a recessive mutation could be responsible for what happened to the children, since their family tree had a history of closely related ancestors while the parents themselves seemed perfectly healthy. But their initial sweep failed to turn up potential clues. Tragically, two more children in the family would later die of sudden cardiac arrest as well, six and eight years after the first deaths, respectively. All of them, no younger than 12, had been playing or exercising right before their deaths. By the time of these newer deaths, though, genetic technology had advanced enough for the team to try looking again. In particular, they were now able to scan a person’s entire exome, the bits of DNA that actually program our cells to make the building block proteins we need to live. And this time, they found a likely suspect: a duplication of DNA found in segments of the RYR2 gene as well as in another region that controls its expression.  The RYR2 gene helps regulate our heart muscle’s calcium release channels (CRC). These channels need to carefully manage the flow of calcium in and out of heart cells to keep the organ healthy and beating as it should during times of rest and stress alike. People are already known to have genetic mutations that can leave them with overactive CRCs—a condition that also raises their risk of sudden cardiac death. But this specific mutation seems to create the opposite problem, leaving victims with too few CRCs. As the team theorized, the children who had died all had two copies of the mutation, while the parents and unaffected siblings all had either one or no copies. They then came across a second large Amish family, unrelated to the first, that also had a history of healthy young people suddenly dying of or barely surviving cardiac arrest. And when the second family was tested, nearly all of those with two copies of the mutated gene had died or developed these symptoms. The mutation and the condition it causes—coined “calcium release channel deficiency syndrome” by the team—still needs to be studied by other researchers before it can be confirmed as a genuine disorder. But so far, 23 people have been identified with the mutation, with 18 having died, across the two families, while more relatives are being tested by the team. Ackerman said his team’s work has been greatly appreciated and celebrated by the families. “The power of closure (figuring out the truth about what was behind all of these tragedies) and clarity (being able to figure out who does and who does not have these markers) is incredible, as you can imagine,” he said. Our genes usually influence our health in very subtle ways. Even people who have a clearly troublesome mutation don’t always become seriously sick. But conventional tests haven’t been able to tell when someone with the condition will have heart troubles. And given how quickly lethal it can be, Ackerman expects that affected individuals will need an implantable cardioverter-defibrillator that can intervene when the heart loses control of itself. More importantly, though, we can now find these people before it’s too late. “Although we could not save the lives of these precious children and teenagers and young adults, we now have a diagnostic biomarker such that no more deaths from CRC deficiency syndrome should have to ever occur again,” Ackerman said. |
|
|
|
Post by Admin on Jan 10, 2020 2:37:00 GMT
Characterization of a Novel Homozygous Multi-exon Ryr2 Duplication Associated With Exertion-related Sudden Unexplained Death in the Young in the Amish Community Using Patient-specific Hipsc-cardiomyocytes David J Tester, CS John Kim, Samantha K Hamrick, Hannah M Bombei, Kristi K Fitzgerald, Carla M Haglund-Turnquist, Dianne L Atkins, Luis A Ochoa, Ian H Law, Joel Temple, Michael J Ackerman Originally published11 Nov 2019Circulation. 2019;140:A12295 Abstract Background: We identified a novel homozygous duplication involving the promoter region and exons 1-4 of RYR2 that is responsible for highly penetrant, exertion-related sudden deaths in the young (SUDY) and sudden cardiac arrests (SCA) in the Amish community without an overt phenotype to suggest RYR2-mediated CPVT. Here, we characterize patient-derived induced pluripotent stem cell--cardiomyocytes (iPSC-CMs) from two unrelated patients.  Methods: Homozygous RYR2-duplication (RYR2-DUP) iPSC lines were generated from two unrelated patients (12-year-old female and a 10-year-old male) with exertional SCA. Two unrelated wild-type (WT) control iPSC lines were analyzed also. Conventional cardiac differentiation methods were used to generate iPSC-CMs. Calcium handling under baseline, 10mM caffeine, and 100nM isoproterenol conditions was assessed by live cell imaging with a Fluo-4 calcium indicator. qRT-PCR, western blot, and immunostaining were used to assess RYR2 gene/RyR2 protein expression. Arrhythmic activity was assessed using an xCELLigence® RTCA CardioECR instrument. Results: There was no difference in baseline Ca2+ handling (Ca2+ transient amplitude, time-to-peak, or decay time) measurements between WT- and the RYR2-DUP-iPSC-CMs patient lines. However, compared to WT-iPSC-CMs, both patient lines demonstrated a dramatic reduction in caffeine and isoproterenol stimulated Ca2+ transient amplitude, suggesting RYR2 loss-of-function. There was a >50% reduction in RYR2 transcript/RyR2 protein expression in both patient iPSC-CMs compared to WT. Delayed afterdepolarization-like ectopy was observed in the RYR2-DUP-iPSC-CMs but not in the WT-iPSC-CMs. Conclusion: Here, we characterized the first iPSC-CM model of a homozygous multi-exon RYR2 duplication mutation identified in two large Amish pedigrees associated with exertion-related SUDY/SCA. Unlike the typical gain-of-function mechanism observed in RYR2-mediated CPVT, the homozygous multi-exon duplication precipitates a loss-of-function in calcium handling, presumably through RYR2 haploinsufficiency. Footnotes For author disclosure information, please visit the AHA Scientific Sessions 2019 Online Program Planner and search for the abstract title. |
|
|
|
Post by Admin on Jan 10, 2020 18:13:12 GMT
Identification of a Novel Homozygous Multi-Exon Duplication in RYR2 Among Children With Exertion-Related Unexplained Sudden Deaths in the Amish Community
David J. Tester, BS1,2,3; Hannah M. Bombei, MS, CGC4; Kristi K. Fitzgerald, MS5; et alJohn R. Giudicessi, MD, PhD1,2,3; Beth A. Pitel, MS6,7; Erik C. Thorland, PhD6,7; Barbara G. Russell, RN-C, BS8; Samantha K. Hamrick, BS1,2,3; C. S. John Kim, PhD1,2,3; Carla M. Haglund-Turnquist1,2,3; Christopher L. Johnsrude, MD8; Dianne L. Atkins, MD4; Luis A. Ochoa Nunez, MD4; Ian Law, MD4; Joel Temple, MD5; Michael J. Ackerman, MD, PhD1,2,3
JAMA Cardiol. Published online January 8, 2020. doi:10.1001/jamacardio.2019.5400
Key Points
Question What is the underlying genetic cause of multiple sudden deaths in young individuals and sudden cardiac arrests observed in 2 large Amish extended families?
Findings In this molecular autopsy and genetic analysis, a novel homozygous multiexon duplication in RYR2 was identified among young Amish individuals with exertion-related sudden deaths and sudden cardiac arrests without an overt phenotype to suggest RYR2-mediated catecholaminergic polymorphic ventricular tachycardia.
Meaning Considering that no cardiac tests reliably identify at-risk individuals, and given the high rate of consanguinity in Amish families, identification of unaffected heterozygous carriers may provide potentially lifesaving premarital counseling and reproductive planning.
Abstract
Importance The exome molecular autopsy may elucidate a pathogenic substrate for sudden unexplained death.
Objective To investigate the underlying cause of multiple sudden deaths in young individuals and sudden cardiac arrests that occurred in 2 large Amish families.
Design, Setting, and Participants Two large extended Amish families with multiple sudden deaths in young individuals and sudden cardiac arrests were included in the study. A recessive inheritance pattern was suggested based on an extended family history of sudden deaths in young individuals and sudden cardiac arrests, despite unaffected parents. A family with exercise-associated sudden deaths in young individuals occurring in 4 siblings was referred for postmortem genetic testing using an exome molecular autopsy. Copy number variant (CNV) analysis was performed on exome data using PatternCNV. Chromosomal microarray validated the CNV identified. The nucleotide break points of the CNV were determined by mate-pair sequencing. Samples were collected for this study between November 2004 and June 2019.
Main Outcomes and Measures The identification of an underlying genetic cause for sudden deaths in young individuals and sudden cardiac arrests consistent with the recessive inheritance pattern observed in the families.
Results A homozygous duplication, involving approximately 26 000 base pairs of intergenic sequence, RYR2’s 5′UTR/promoter region, and exons 1 through 4 of RYR2, was identified in all 4 siblings of a family. Multiple distantly related relatives experiencing exertion-related sudden cardiac arrest also had the identical RYR2 homozygous duplication. A second, unrelated family with multiple exertion-related sudden deaths and sudden cardiac arrests in young individuals, with the same homozygous duplication, was identified. Several living, homozygous duplication–positive symptomatic patients from both families had nondiagnostic cardiologic testing, with only occasional ventricular ectopy occurring during exercise stress tests.
Conclusions and Relevance In this analysis, we identified a novel, highly penetrant, homozygous multiexon duplication in RYR2 among Amish youths with exertion-related sudden death and sudden cardiac arrest but without an overt phenotype that is distinct from RYR2-mediated catecholaminergic polymorphic ventricular tachycardia. Considering that no cardiac tests reliably identify at-risk individuals and given the high rate of consanguinity in Amish families, identification of unaffected heterozygous carriers may provide potentially lifesaving premarital counseling and reproductive planning.
|
|
|
|
Post by Admin on Feb 9, 2020 21:34:13 GMT
As our primary interface with the environment, the immune system is thought to have evolved under strong selective pressure from pathogens (Barreiro and Quintana-Murci, 2010, Fumagalli et al., 2011, Karlsson et al., 2014). When human populations migrated out of Africa, they encountered markedly different pathogenic environments, likely resulting in population-specific selection on the immune response (Barreiro and Quintana-Murci, 2010, Fumagalli et al., 2011, Karlsson et al., 2014). Substantial evidence supports this hypothesis at the genetic level. However, we still know little about the extent to which neutral or adaptive inter-population genetic differences affect the actual immune response to pathogens. 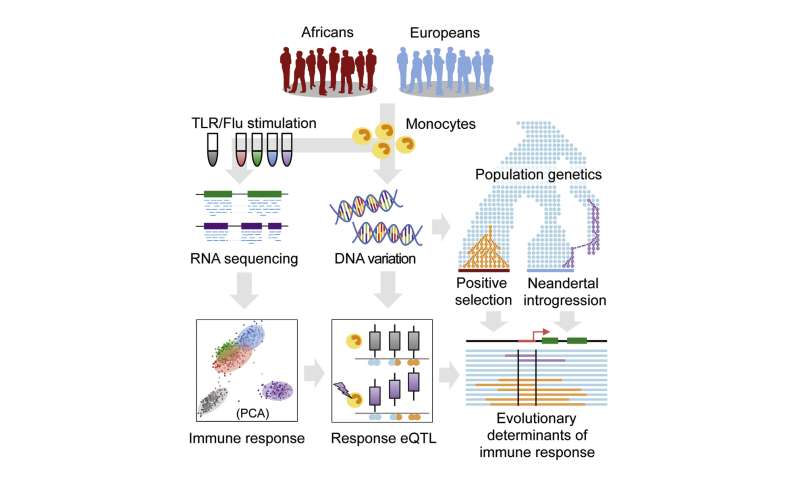 Addressing this gap is not only important for understanding recent human evolution, but may also help reveal the molecular basis of ancestry-related differences in disease susceptibility. Individuals from different populations vary considerably in their susceptibility to many infectious diseases, chronic inflammatory disorders, and autoimmune disorders. For tuberculosis, systemic lupus erythematosus, systemic sclerosis, psoriasis, and septicemia, African American (AA) and European American (EA) individuals exhibit an up to 3-fold difference in prevalence (reviewed in Brinkworth and Barreiro, 2014, Pennington et al., 2009, Richardus and Kunst, 2001). These observations argue in favor of significant ancestry-related differences in immune response, especially in susceptibility to inflammation (Pennington et al., 2009, Richardus and Kunst, 2001). Such differences almost certainly involve major contributions from the environment. However, genome-wide association studies (GWAS) also support a key role for genetic factors, as many of the GWAS-variants associated with infectious, autoimmune, and inflammatory diseases present extreme differences in allele frequency (Fst > 0.4) between human populations, again supporting a possible history of population-specific selection (Brinkworth and Barreiro, 2014). GWAS results also indicate that susceptibility to many common immune-related diseases is primarily controlled by non-coding variants (Gusev et al., 2014, Hindorff et al., 2009, Schaub et al., 2012). Thus, many ancestry-related differences in disease susceptibility may result from genetically controlled transcriptional differences in immune responses to inflammatory signals. This idea is consistent with recent expression quantitative trait locus (eQTL) mapping studies in innate immune cells exposed to immune antigens or live infectious agents (Barreiro et al., 2012, Çalışkan et al., 2015, Fairfax et al., 2014, Lee et al., 2014). Such immune “response eQTL” studies have identified hundreds of genetic variants that both explain variation in the host immune response and are significantly enriched among GWAS-associated loci. However, because studies to date have mostly focused on individuals of European ancestry, the degree to which such variants contribute to population differences in the immune response remains unclear. Here, we report an RNA-sequencing (RNA-seq)-based immune response eQTL study to test for the effects of African versus European ancestry on the transcriptional response to several live bacterial pathogens. We integrate statistical and evolutionary genetic analyses with primary macrophage gene expression levels, before and after infection, to characterize ancestry-related differences in the immune response. Our analyses address three fundamental questions about recent evolution in the human immune system: (1) the degree to which innate immune responses are differentiated by European versus African ancestry, (2) the genetic variants that account for such differences, and (3) the evolutionary mechanisms (neutral genetic drift versus positive selection) that led to their establishment in modern human populations. |
|
|
|
Post by Admin on Mar 18, 2020 21:35:27 GMT
The Genetic Legacy of the Expansion of Turkic-Speaking Nomads across Eurasia Bayazit Yunusbayev, Mait Metspalu, [...], and Richard Villems Abstract The Turkic peoples represent a diverse collection of ethnic groups defined by the Turkic languages. These groups have dispersed across a vast area, including Siberia, Northwest China, Central Asia, East Europe, the Caucasus, Anatolia, the Middle East, and Afghanistan. The origin and early dispersal history of the Turkic peoples is disputed, with candidates for their ancient homeland ranging from the Transcaspian steppe to Manchuria in Northeast Asia. Previous genetic studies have not identified a clear-cut unifying genetic signal for the Turkic peoples, which lends support for language replacement rather than demic diffusion as the model for the Turkic language’s expansion. We addressed the genetic origin of 373 individuals from 22 Turkic-speaking populations, representing their current geographic range, by analyzing genome-wide high-density genotype data. In agreement with the elite dominance model of language expansion most of the Turkic peoples studied genetically resemble their geographic neighbors. However, western Turkic peoples sampled across West Eurasia shared an excess of long chromosomal tracts that are identical by descent (IBD) with populations from present-day South Siberia and Mongolia (SSM), an area where historians center a series of early Turkic and non-Turkic steppe polities. While SSM matching IBD tracts (> 1cM) are also observed in non-Turkic populations, Turkic peoples demonstrate a higher percentage of such tracts (p-values ≤ 0.01) compared to their non-Turkic neighbors. Finally, we used the ALDER method and inferred admixture dates (~9th–17th centuries) that overlap with the Turkic migrations of the 5th–16th centuries. Thus, our results indicate historical admixture among Turkic peoples, and the recent shared ancestry with modern populations in SSM supports one of the hypothesized homelands for their nomadic Turkic and related Mongolic ancestors. Author Summary Centuries of nomadic migrations have ultimately resulted in the distribution of Turkic languages over a large area ranging from Siberia, across Central Asia to Eastern Europe and the Middle East. Despite the profound cultural impact left by these nomadic peoples, little is known about their prehistoric origins. Moreover, because contemporary Turkic speakers tend to genetically resemble their geographic neighbors, it is not clear whether their nomadic ancestors left an identifiable genetic trace. In this study, we show that Turkic-speaking peoples sampled across the Middle East, Caucasus, East Europe, and Central Asia share varying proportions of Asian ancestry that originate in a single area, southern Siberia and Mongolia. Mongolic- and Turkic-speaking populations from this area bear an unusually high number of long chromosomal tracts that are identical by descent with Turkic peoples from across west Eurasia. Admixture induced linkage disequilibrium decay across chromosomes in these populations indicates that admixture occurred during the 9th–17th centuries, in agreement with the historically recorded Turkic nomadic migrations and later Mongol expansion. Thus, our findings reveal genetic traces of recent large-scale nomadic migrations and map their source to a previously hypothesized area of Mongolia and southern Siberia.  Fig 1 Geographic map of samples included in this study and linguistic tree of Turkic languages. Introduction Linguistic relatedness is frequently used to inform genetic studies [1] and here we take this path to reconstruct aspects of a major and relatively recent demographic event, the expansion of nomadic Turkic-speaking peoples, who reshaped much of the West Eurasian ethno-linguistic landscape in the last two millennia. Modern Turkic-speaking populations are a largely settled people; they number over 170 million across Eurasia and, following a period of migrations spanning the ~5th–16th centuries, have a wide geographic dispersal, encompassing Eastern Europe, Middle East, Northern Caucasus, Central Asia, Southern Siberia, Northern China, and Northeastern Siberia [2–4]. The extant variety of Turkic languages spoken over this vast geographic span reflects only the recent (2100–2300 years) history of divergence, which includes a major split into Oghur (or Bolgar) and Common Turkic [5, 6]. This period was preceded by early Ancient Turkic, for which there is no historical data, and a long-lasting proto-Turkic stage, provided there was a Turkic-Mongolian linguistic unity (protolanguage) around 4500–4000 BCE [7, 8]. The earliest Turkic ruled polities (between the 6th and 9th centuries) were centered in what is now Mongolia, northern China, and southern Siberia. Accordingly, this region has been put forward as the point of origin for the dispersal of Turkic-speaking pastoral nomads [3, 4]. We designate it here as an “Inner Asian Homeland” (IAH) and note at least two issues with this working hypothesis. First, the same approximate area was earlier dominated by the Xiongnu Empire (Hsiung-nu) (200 BCE–100 CE) and later by the short-lived Xianbei (Hsien-pi) Confederation (100–200 CE) and Rouran State (aka Juan-juan or Asian Avar) (400–500 CE). These steppe polities were likely established by non-Turkic-speaking peoples and presumably united ethnically diverse tribes. It is only in the second half of the 6th century that Turkic-speaking peoples gained control of the region and formed the rapidly expanding Göktürk Khaganate, succeeded soon by numerous khanates and khaganates extending from northeastern China to the Pontic-Caspian steppes in Europe [2–4]. Secondly, Göktürks represent the earliest known ethnic unit whereby Turkic peoples appear under the name Turk. Yet, Turkic-speaking peoples appear in written historical sources before that time, namely when Oghuric Turkic-speaking tribes appear in the Northern Pontic steppes in the 5th century, much earlier than the rise of Göktürk Khaganate in the IAH[9]. Thus, the early stages of Turkic dispersal remain poorly understood and our knowledge about their ancient habitat remains a working hypothesis. Previous studies based on Y chromosome, mitochondrial DNA (mtDNA), and autosomal markers show that while the Turkic peoples from West Asia (Anatolian Turks and Azeris) and Eastern Europe (Gagauzes, Tatars, Chuvashes, and Bashkirs) are generally genetically similar to their geographic neighbors, they do display a minor share of both mtDNA and Y haplogroups otherwise characteristic of East Asia [10–15]. Expectedly, the Central Asian Turkic speakers (Kyrgyz, Kazakhs, Uzbeks, and Turkmens), share more of their uniparental gene pool (9–76% of Y chromosome and over 30% of mtDNA lineages) with East Asian and Siberian populations [16, 17]. In this regard, they differ from their southern non-Turkic neighbors, including Tajiks, Iranians, and different ethnic groups in Pakistan, except Hazara. However, these studies do not aim to identify the precise geographic source and the time of arrival or admixture of the East Eurasian genes among the contemporary Turkic-speaking peoples. The “eastern” mtDNA and even more so Y-chromosome lineages (given the resolution available to the studies at the time) lack the geographic specificity to explicitly distinguish between regions within Northeast Asia and Siberia, and/or Turkic and non-Turkic speakers of the region [18, 19]. Several studies using genome-wide SNP panel data describe the genetic structure of populations in Eurasia and although some include different Turkic populations [15, 20–24], they do not focus on elucidating the demographic past of the Turkic-speaking continuum. In cases where more than one geographic neighbor is available for comparison, Turkic-speaking peoples are genetically close to their non-Turkic geographic neighbors in Anatolia [22, 25], the Caucasus [15], and Siberia [21, 23]. A recent survey of worldwide populations revealed a recent (13th–14th century) admixture signal among the three Turkic populations (Turks, Uzbeks, and Uygurs) and one non-Turkic population (Lezgins) with Mongolas (from northern China), the Daurs (speaking Mongolic language), and Hazaras (of Mongol origin) [26]. This study also showed evidence for admixture (dating to the pre-Mongol period of 440–1080 CE) among non-Turkic (except Chuvashes) East European and Balkan populations with the source group related to modern Oroqens, Mongolas, and Yakuts. This is the first genetic evidence of historical gene flow from a North Chinese and Siberian source into some north and central Eurasian populations, but it is not clear whether this admixture signal applies to other Turkic populations across West Eurasia. Here we ask whether it is possible to identify explicit genetic signal(s) shared by all Turkic peoples that have likely descended from putative prehistoric nomadic Turks. Specifically, we test whether different Turkic peoples share genetic heritage that can be traced back to the hypothesized IAH. More specifically, we ask whether this shared ancestry occurred within an historical time frame, testified by an excess of long chromosomal tracts identical by descent between Turkic-speaking peoples across West Eurasia and those inhabiting the IAH. To address these questions we used a genome-wide high-density genotyping array to generate data on Turkic-speaking peoples representing all major branches of the language family (Fig 1B). |
|

