|
|
Post by Admin on Aug 17, 2020 20:44:44 GMT
Autosomal SNP variance pattern in the native populations of Sakha and beyond Analyses of more than 600,000 common single nucleotide polymorphism loci from the nuclear genome were based on a sample of 758 individuals from 55 populations. The sample set includes 40 previously unpublished samples from Siberia and compatible published data from Europe, Asia and America [36-38] to represent the regional context (see Additional file 10). In the heatmap plot of FST distances (Figure 4), populations from Sakha form a cluster of low genetic distance, with Yakuts and Evenks being the closest. The smooth transition from Sakha to South Siberia and on to East Asia contrasts with the discontinuity between Sakha and Northeast Siberia. 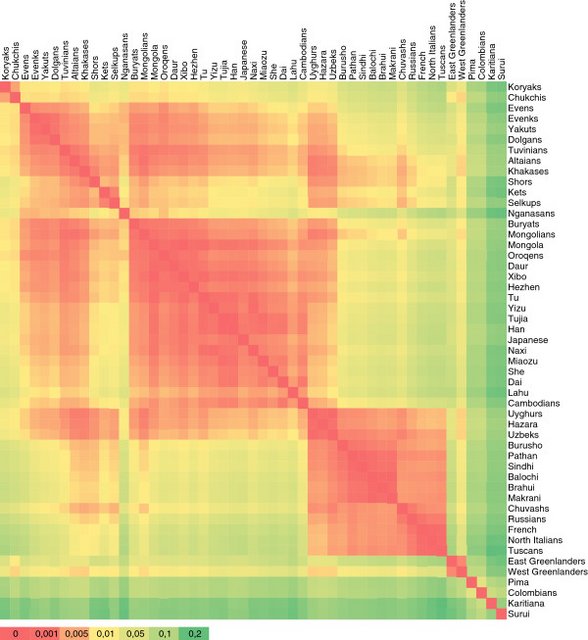 Figure 4 Heatmap plot of FST distances. Only populations with N≥4 are included (see Additional file 10 for list of samples). The PC analysis demonstrates the clustering of populations from Sakha close to Tuvinians and Buryats from southern Siberia as well as to Mongolians from Mongolia, whereas they are separated from the neighbouring Chukchi and Koryaks (Figure 5). It is remarkable that Nganasans who live in relative isolation on the Taymyr Peninsula are situated near the populations of Sakha in the PC plot. Evenks and Yakuts together with some Dolgans, Evens and Yukaghirs cluster close together, while the rest of the Dolgans, Yukaghirs and a few Yakuts are dispersed between Siberian/Central Asian and one Yukaghir even among East European populations. The majority of Evens form a distinct cluster near the Yakut/Evenk and Nganasan groups. 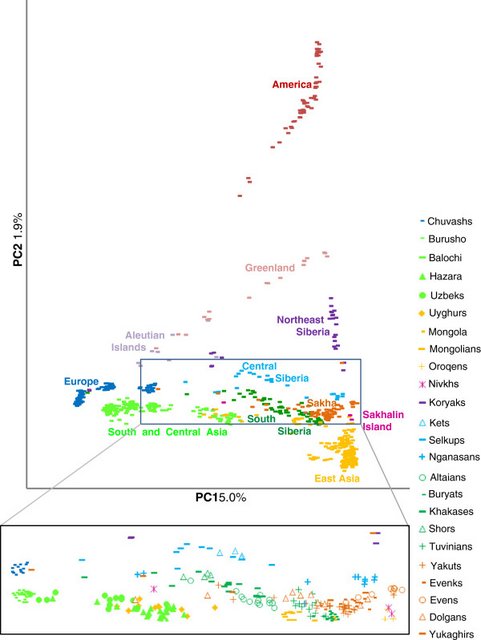 Figure 5 PCA of the native populations of Sakha in the context of other Eurasian and American populations. In order to study the genetic relationships of the populations in more detail, we used the model-based algorithm ADMIXTURE [50] that computes quantitative estimates for inferring the ancestry of individuals from K number of constructed ancestral populations. As expected at K=3 Siberian and East Asian ancestry palettes are largely indistinguishable (Figure 6, see Additional file 11 and Additional file 12). However, there is a minor signal that unites Siberians with Amerinds and Greenlanders, probably reflecting deep-rooted shared ancestry of Siberians and Native Americans. As previously shown [36,37], starting from certain K values (here at K=4), the dominant East Asian – Siberian ancestry component (yellow) splits into two. Lemon yellow is present among northern Asian populations as well as Greenlanders and Aleuts and dark yellow is characteristic of Hans and southern Chinese populations. At K=6, the lemon yellow splits further to discriminate the Greenland and Aleutian populations from the Siberians. Importantly, this new component (olive) is also present among the Chukchis and Koryaks, likely testifying to some level of Beringian continuity. Starting from K=8, Greenlanders acquire their own color, largely restricted to them only. At K=13, a new ancestry component (lavender) appears among the Siberian populations. It is most prominently apparent among the Nganasans and is present as a minor signal in almost all Siberian and northern Chinese populations. Although the level of variation in log-likelihood scores (LLs) within a fraction (10%) of runs with the highest LLs shows that the global maximum is probably not reached at K = 13, seven runs from ten exhibit this new ancestry component (see Additional file 12 and Additional file 13). In the Siberian ancestry palette there is another minor signal, explained by an overlap with the major component in European populations. This component, accounting for about 20% of the ancestry of individuals from South Siberia, decreases practically to zero in northern Siberian populations. However, a few individuals from small northern populations (and, incidentally, all of the Aleuts in our sample) show an exceptionally high proportion of the European component. |
|
|
|
Post by Admin on Aug 18, 2020 20:22:38 GMT
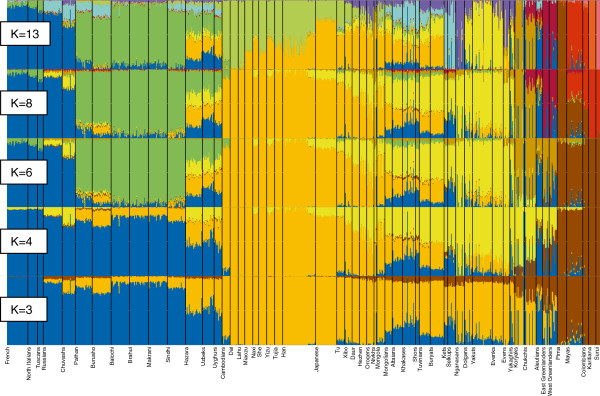 Figure 6 ADMIXTURE plots. Ancestry proportions of the 758 individuals studied (from 55 populations) as revealed by the ADMIXTURE software at K = 3, K = 4, K = 6, K = 8, and K = 13. The main feature of the autosomal admixture pattern of the native populations of Sakha is the prevalence of the lemon yellow component (Figure 6), especially in Yakuts and, in a smaller extent, in Evenks and Dolgans. The second Siberian-specific component (lavender) makes up approximately two third of the ancestry of Evens, but is barely present in Yakuts. Evens and Yukaghirs show a minor signal overlapping with the dominant component in Koryaks and Chukchis. It most probably reflects a limited gene flow from Northeast Siberia. The presence of the “European” (blue) component in the native populations of Sakha likely testifies to admixture that is recent and/or ongoing, or has been considerable in the past. Although the ancestry panels do not offer specific time estimates for putative admixture events, it is reasonable to suggest that “old” admixture or shared ancestry would even out within a population, whereas more recent and ongoing admixture events would exhibit with large differences between individuals in a population. The latter can most obviously be seen among the Yukaghirs. Mantel correlation tests Mantel tests were used to clarify the role of geography and linguistic diversity in the formation of the genetic variation spatial patterns in northern Asia. We started by analyzing the Altaic-speaking populations, the most numerous and widespread populations in northern Asia. The term “Altaic” has been used here to denote a language family that includes the Tungusic, Turkic and Mongolic languages [51]. A Mantel test performed on 18 Altaic-speaking populations from Siberia, Central Asia, Mongolia and northern China (see Additional file 10) revealed a statistically significant positive correlation between autosomal SNP variation and geography (r = 0.68, P < 0.001) (see Additional file 14). Geography explains 46.8% of the genetic variance. Additional Mantel tests applied to mtDNA and Y chromosome data of 22 Altaic-speaking populations (see Additional file 15) show a correlation between mtDNA variation and geography (r = 0.55, P < 0.001), whereas Y-chromosome variation exhibits a weak partial correlation with linguistic data (r = 0.18, P = 0.014) (see Additional file 14). Isolation by distance is the most plausible explanation for the positive correlation between maternal genetics and geography. Patrilocality and a patrilineal clan structure combined with strict exogamy common among Altaic-speaking populations [52] could cause a greater degree of female than male admixture and/or the adoption of languages by females to a greater extent than by males. The correlation between paternal genetics and linguistics hints at the concordant spread of genes and languages, but this has not been the sole process, because linguistic data explain only 3.3% of the Y-chromosome variance. When the Chukchis and Koryaks, representatives of the Chukotko-Kamchatkan language family from Northeast Siberia, were added to the analysis, a considerable partial correlation between autosomal and linguistic variation appeared (r = 0.55, P = 0.002) in addition to the correlation between genetics and geography. Both linguistics and geography explain approximately one third of the genetic variance. The results of the Mantel test based on mtDNA data are very similar (see Additional file 14). In contrast, there is no correlation between paternal genetics and geographic distances or linguistic diversity. Go to: Discussion In the following section, we discuss (1) the role of South Siberia in the formation of the existing genetic variation in Sakha, (2) the genetic discontinuity between Sakha and Northeast Siberia, and (3) the origin of the West Eurasian component in the gene pool of the native populations of Sakha. Sakha as an extension of South Siberia The PCA plot and FST values based on autosomal SNP data demonstrate the genetic proximity of the populations of Sakha to the geographically and linguistically neighboring populations from South Siberia, particularly Tuvinians and Buryats. The ADMIXTURE analysis reveals that the genetic heritage of the populations of Sakha in its great majority is characterized by Siberian-specific ancestry components which stem from East Asia. The Siberian components, comprising the component denoted by lemon yellow that achieves its highest proportion in Yakuts and the one denoted by lavender represented most clearly in Evens, connect the inhabitants of Sakha with southern Siberian populations. Similarly, phylogenetic analyses of uniparental data show that East Asian mtDNA and Y-chromosome haplogroups form the main part of Sakha gene pool, and the majority of maternal and paternal lineages found in Sakha (Figure 1 and and2)2) are nested in the larger genetic variation of South Siberia [19,20,23,46]. In addition, a fair number of mtDNA haplogroups (C4a1c, C4a1d, C4a2, C4b1, C4b3, C5b1a, D4i2, D4j2, D4j4, D4j5, D4j8, D4o2) as well as Y-chromosome haplogroups C3c and N1b common in Sakha have been dated to the Siberian Neolithic [20,23,35,46], the beginning of which coincided with the period of climatic optimum in the Postglacial. These results all suggest that a substantial part of maternal and paternal lineages in Sakha might stem from Neolithic expansions in South Siberia, having been carried to Sakha afterwards at different times by different peoples. Y-chromosome haplogroup C3c, the most frequent among Evenks and Evens in Sakha (Figure 2, see Additional file 5), might have arrived with the ancestors of the Tungusic peoples. This scenario is supported by the observation that C3c is much more frequent among various Tungusic-speaking peoples from Siberia and northern China than in their neighbors [47]. MtDNA haplogroups Z1a and C4b could represent traces of more ancient migrations in northern Siberia, as these haplogroups have been dated as older and, furthermore, their known sub-clades are found almost exclusively in Arctic populations (see Additional file 2 and Additional file 3) [20,35]. Z1a stretches all over Siberia, but three distinct sub-clades (Z1a1b, Z1a2a and Z1a3) are represented mainly in the northern regions. Interestingly, all three clades encompass the Yukaghirs – nowadays a very small population residing in northeastern Sakha and Chukotka. Z1a1b includes Nganasans from the Taymyr Peninsula and Evens from Sakha besides the Yukaghirs. Analyses of autosomal data confirm the genetic relatedness of Nganasans, Yukaghirs and Evens (Figures 5 and and6).6). These results support the scenario that the ancestors of Yukaghirs originate in the Taymyr Peninsula in Neolithic times. In the middle of the second millennium BC, the Yukaghir ancestors spread from the Taymyr Peninsula to the east, under pressure from immigrating groups [10]. In the first half of the second millennium AD, the expansion of Evenks cut the Yukaghirs off from Samoyedic-speaking groups and forced them further east, where they ended up being surrounded by the Chukchis, Koryaks, Evens and the ancestors of Yakuts. Remarkable gene flow between the Yukaghirs and Evens is revealed by mtDNA analysis – in our sample, 71% of maternal lineages are shared between the Yukaghirs and Evens. In addition, the Yukaghirs have acquired a few maternal lineages (Z1a2a, G1b, C5a2a) from the Koryaks. The clearest and most abundant traces in the Sakha gene pool have been left by the most recent migrations. The prevailing Y-chromosome haplogroup among the Yakuts, N1c, makes up over two thirds of paternal lineages in our sample. The Yakut-specific branch comprises also lineages from Evenks, Evens and Dolgans, most probably due to male gene flow from the Yakuts. The fact that the ancestral STR haplotype of the Yakut-specific clade is present among the Tuvinians, Tofalars and Sojots from the eastern Sayan regions [46] could point to the putative “homeland” of Yakutian N1c predecessors. In addition, at least one maternal lineage, the sub-clade of C4a1c defined by the back-mutation C16298T, may have migrated together with the Y-chromosome haplogroup N1c. This clade, represented in contemporary (see Additional file 1) as well as ancient Yakuts [43], has also been found among the Tuvinians [19]. Close similarities between the Yakut and Turkic languages spoken in the Altai-Sayan region [53], as well as some other aspects of the Yakut culture (e.g., pastoralism, clothing, festivals) [54], point to ancestral ties between the Yakuts and the southern Turkic peoples. These facts suggest that the Yakuts originate from the Altay-Sayan region. On the other hand, some mtDNA (D5a2a2, M13a1b, A8, G2a5) and Y-chromosome haplogroups (C3d, the Buryat-specific cluster of N1c) in the Yakut gene pool are shared with Mongolic populations (Buryats, Khamnigans, Mongolians) from the Baikal area and Mongolia [20,21,23,46]. This fact is consistent with the hypothesis proposed on the basis of archaeological findings, postulating that the Yakuts originate from the ancient Turkic-speaking Kurykan people from the Lake Baikal region [5,10,11]. Estimates of the expansion time of the Yakut-specific clade of Y-chromosome haplogroup N1c support the hypothesis: the clade started to diversify ~1.6 kya, about the same time (6th – 10th century AD) when the culture of the Kurykans flourished on the shores of Lake Baikal. The second expansion of the clade, dated to ~0.9 kya, might coincide with the migration of the Yakuts´ ancestors to the middle reaches of the Lena River in the 11th-13th centuries AD. Thus, taking into account the aforementioned facts, the Yakut ancestors most probably originated from the Altay-Sayan region and settled for a time in the Lake Baikal area before migrating northwards along the Lena River. |
|
|
|
Post by Admin on Aug 19, 2020 6:31:46 GMT
Genetic discontinuity between Sakha and Northeast Siberia
Although the genetic heritage of the native populations from Sakha as well as Northeast Siberia lies in its great majority in the common East Asian gene pool, analyses of autosomal as well as uniparental markers revealed the genetic divergence between these neighboring regions. The core of the mtDNA gene pool of the native people of Sakha consists of a number of haplogroup C and D sub-lineages (see Additional file 1). The Koryaks and Chukchis harbor only a few topmost clades of these two haplogroups (C4b2, C5a2a, D4b1a2a and D2a) [30,39], but these lineages are uncommon or even absent in Sakha. The prevailing haplogroups among Chukchis – A2a, A2b and D2a [30] – were not found in Sakha, while G1b, Y1a and Z1a2, common among the Koryaks [39], are present in Sakha at low frequencies. The Koryak-specific maternal lineages found in Sakha most probably suggest a recent limited maternal gene flow from Northeast Siberia. In contrast, the Y-chromosome haplogroups N1c and C3 prevalent in Sakha (Figure 2) are also represented in Northeast Siberia [47,55]. However, the Koryaks [23] and the populations of Sakha (see Additional file 5) do not share Y-STR haplotypes of haplogroup C3, and the N1c haplotypes detected among the Koryaks and Chukchis are quite distinct from those of other Siberians [22,46]. In addition, the presence of the Y-chromosome haplogroup Q1a3a [Q(M3)] among the Chukchis [55] distinguishes them from the populations of Sakha, where this haplogroup has not been found. These results point not so much to recent male-mediated gene flow between these neighboring regions but rather to multiple separate migrations from the same source area. The PCA plot (Figure 5) and FST values (Figure 4) based on autosomal SNP data also show that the Koryaks and Chukchis are distant from the populations of Sakha. However, ADMIXTURE analysis reveals also a deep-rooted shared ancestry of the inhabitants of Sakha and extreme Northeast Siberia (Figure 6). The genetic data are in good accordance with archaeological findings that demonstrate direct cultural contacts between Kamchatka, Chukotka and Yakutia during the late Paleolithic and Neolithic, and suggest a period of relative isolation for the extreme Northeast only since the second – first millennia B.C. [56].
The strong positive partial correlation between genetic and linguistic variation shown by the Mantel test (see Additional file 14) suggests that the same past population processes have shaped linguistic as well as genetic divergence between Sakha and the Kamchatka-Chukotka region. The present study, as well as previous ones [26,29,43], have revealed the main features of the gene pool of the native populations of Sakha to have been shaped by migrations from South Siberia, in particular by relatively recent expansions of Tungusic- and Turkic-speaking peoples. The Chukchis and Koryaks, inhabitants of the Kamchatka-Chukotka region, are considered to be the descendants of the Neolithic indigenous people of Northeast Siberia [56]. The Chukchi and Koryak languages, together with Kerek and Alutor, form the closely-knit Chukotkan group in the Chukotko-Kamchatkan language family [57], the speakers of which inhabit extreme Northeast Siberia. The Chukotko-Kamchatkan languages have no generally accepted relation to any other language family, but sometimes they are classified together with the Nivkh, Yukaghir and Yeniseian languages among the Paleosiberian languages that are believed to represent a remnant of a much richer linguistic palette of Siberia that existed before the Altaic, Uralic and Indo-European languages expanded across most of Siberia [58]. The prevalent mtDNA haplogroups among the Chukchis – A2a, A2b and D2a [30] – are shared with Greenland Eskimos, Aleuts and a few native populations of North America [15,33,59,60] due to either recent female-mediated gene flow from Eskimos/Aleuts or deep shared ancestry. Similarly, analysis of autosomal SNP data reveals an ancestry component shared between the Chukchis and Greenland Eskimos (Figure 6). MtDNA haplogroups G1b and Y1a, common among the Koryaks [39], and G1b, common among the Chukchis [30], connect them with the Nivkhs from the Sakhalin Island [34]. ADMIXTURE analysis also points to genetic connections between the Chukchis, Koryaks and Nivkhs (Figure 6). In addition, quite a strong relationship between the Nivkh and Chukotko-Kamchatkan languages has been shown [57]. MtDNA haplogroups C4b2 and C5a2a form a part of the “C world” common in South Siberia and Sakha, but their autochthonous nature and coalescence time (~1.2 kya and ~2.6 kya, respectively [20]) hint at a period of isolation from the rest of Siberia. Considering the facts discussed above it is probable that large-scale expansions of Tungusic and Turkic peoples in Siberia, which replaced and/or assimilated ancient aboriginal people in Sakha, as well as the relative isolation between Sakha and Northeast Siberia in the subsequent period, are responsible for the formation of a genetic discontinuity between these neighboring regions.
Origin of the West Eurasian genetic component in the gene pool of the native populations of Sakha
Although the genetic heritage of the native populations of Sakha is mostly of East Asian-ancestry, analyses of autosomal SNP data as well as haploid loci also show a minor West Eurasian genetic component. The patchy presence of the “European” (blue) component in the ADMIXTURE plot (Figure 6), most pronounced in Yukaghirs, probably testifies to recent admixture with Europeans. In addition, the presence of European-specific paternal lineages R1a-M458, I1 and I2a among Yakuts, Dolgans, Evenks and Yukaghirs likely points to a recent gene flow from East Europeans. Although only individuals with self-reported un-admixed ancestry for at least two generations were included in the study of haploid loci, mistakes in ethnic self-identification cannot be entirely excluded. One of the main sources of gene flow has likely been Russians who accounted for 37.8% of the population of Sakha in 2010 [61]. The migration of Russians (at first mainly men) to eastern Siberia started already in the 17th century, when Yakutia was incorporated into the Russian Empire [62].
The mtDNA haplogroup J detected in the remains from a Yakut burial site dated to the beginning of the 17th century [41], long before the beginning of the settlement of Russian families in the 18th century [63], clearly points to more ancient gene flow from western Eurasia. The presence of haplogroups H8, H20 and HV1a1a among the Yakuts, Dolgans and Evenks (Figure 1) also suggests gene flow other than from Russians, because these haplogroups are rare (H8 and H20) or even absent (HV1a1a) among Russians [64-67], but are common among southern Siberian populations as well as in the Caucasus, the Middle and Near East [19,68-70]. Moreover, the HVSI haplotypes of H8, H20a and HV1a1a in our sample exactly match those in the Buryats from the Buryat Republic [19]. Similarly, the Y-chromosome haplogroup J in Dolgans and Evens very likely testifies to gene flow through South Siberia, as it is present among native South Siberian populations [47,71]. The scenario of ancient gene flow from West Eurasia is supported by ancient DNA data, which show that in the Bronze and Iron Ages, South Siberia, including the Altai region, was an area of overwhelmingly predominant western Eurasian settlement [72,73], and the Indo-European migration even reached northeastern Mongolia [74]. To summarize, the West Eurasian genetic component in Sakha may originate from recent admixture with East Europeans, whereas more ancient gene flow from West Eurasia through Central Asia and South Siberia is also probable.
Conclusions
The analysis of haploid (mtDNA, NRY) and diploid loci of genome provides further evidence that the genetic heritage of indigenous people of Sakha lies in its great majority in the common East Asian gene pool while West Eurasian influence has been minor. The Turkic-speaking Yakuts retain traces of related populations from the Altai-Sayan region as well as distinctive maternal and paternal lineages, either originating from the Mongolic peoples in the Lake Baikal area or influenced by the long timescale residence in Sakha in close proximity to the Tungusic-speaking Evenks. Genetic data fit well with the linguistic and historical evidence regarding the origin of Yakuts. The Yukaghir gene pool harbor traces of more ancient migration(s) in the arctic regions of Siberia, complemented by recent admixture with Europeans. The Evens, linguistically very close to the Evenks, have also acquired genetic inputs from their geographic neighbors, the Yukaghirs. The European component in Sakha may originate from recent admixture with East Europeans and/or from more ancient gene flow from West Eurasia through Central Asia and South Siberia. The genetic proximity of the native populations of Sakha to South Siberians and the genetic divergence between them and the Chukchis and Koryaks, shown by the present study, suggest that the region of Sakha was colonized by multiple migrations from South Siberia with only minor gene flows from the Lower Amur/Southern Okhotsk region and/or Kamchatka.
|
|
|
|
Post by Admin on Dec 29, 2020 5:07:03 GMT
"Super-archaic" gene regions have been passed down to some living people. FOR YEARS, scientists have tried to detangle the actors that played a role in human evolution's complicated story. We know that our modern human ancestors mated with other early hominin groups, such as the Neanderthals and the Denisovans. But further research suggests they may have led a more promiscuous life than previously thought.  In a study published in August, researchers analyzed human DNA using a brand-new method and, in doing so, they were able to unearth parts of the DNA that came from other species. Using a method called the “ancestral recombination graph” algorithm, dubbed ARGweaver-D, the study team discovered that the Denisovans possessed a genome that contained one percent of DNA from an unknown distant relative. The algorithm works by analyzing the genome to identify segments of DNA that may have come from different species, no matter the time and no matter the source. The team applied the algorithm to look at genomes from two Neanderthals, a Denisovan, and four modern humans. Using this specific approach, they were able to look further back in time than any other existing technology has been able to so far.  According to their findings, 3 percent of the Neanderthal genome can be traced back to ancient humans, and this interbreeding is estimated to have occurred between 200,000 and 300,000 years ago. But the researchers found something even more fascinating. We know that the Denisovans, a group of extinct humans who lived in Asia, and Homo sapiens also intermingled. It turns out that, in doing so, they passed on some of the genes of this mysterious ancestor — what the researchers are calling a “super-archaic” population. They found that 1 percent of the Denisovan genome came from this mysterious relative. It's estimated, in turn, some modern humans could hold about 15 percent of these "super-archaic" gene regions. WHO COULD THIS UNKNOWN ANCESTOR BE? One potential contender could be Homo erectus, an extinct human ancestor who lived in Africa about 1 million years ago. The problem is, we’ve never found any H. erectus DNA, so the most we can do is guess at the moment. The findings only further complicate the evolutionary history of the modern human — it's possible there may have been ever more populations involved. |
|
|
|
Post by Admin on Jan 7, 2021 22:20:17 GMT
Human population dynamics and Yersinia pestis in ancient northeast Asia Science Advances 06 Jan 2021: Vol. 7, no. 2, eabc4587 DOI: 10.1126/sciadv.abc4587 Abstract We present genome-wide data from 40 individuals dating to c.16,900 to 550 years ago in northeast Asia. We describe hitherto unknown gene flow and admixture events in the region, revealing a complex population history. While populations east of Lake Baikal remained relatively stable from the Mesolithic to the Bronze Age, those from Yakutia and west of Lake Baikal witnessed major population transformations, from the Late Upper Paleolithic to the Neolithic, and during the Bronze Age, respectively. We further locate the Asian ancestors of Paleo-Inuits, using direct genetic evidence. Last, we report the most northeastern ancient occurrence of the plague-related bacterium, Yersinia pestis. Our findings indicate the highly connected and dynamic nature of northeast Asia populations throughout the Holocene. INTRODUCTION Siberia covers an extensive area in northeast Asia, stretching eastward from the Ural Mountains to the Pacific Ocean and northward from the Altai Saian Mountains to the Arctic Ocean. Despite being one of the most sparsely populated regions on Earth (1), this large territory has an intriguing human history as home to multiple ethnic groups and a source area for the peopling of Americas (2). Anatomically modern humans pioneered the northeastern part of Siberia ~38,000 years ago, in parallel with the dispersal of the parts of megafauna, i.e., mammoth, woolly rhino, and cave lion (3–5). These dispersals took place before the beginning of the Last Glacial Maximum (LGM) ~33,000 years ago. By the time the glaciation came to an end, ~20,000 to 18,000 years ago, both humans and the megafauna in the area had suffered the consequences of the LGM, but new data have contradicted earlier speculation of a marked settlement decline in the area (5–7). Traces of LGM and post-LGM human activity in northeast Asia, as evidenced by the presence of lithic technologies and pre-Neolithic pottery, are found across the east Siberian plateau as well as the Trans-Baikal area, east of Lake Baikal (2, 8–11). Throughout the Holocene, multiple cultural complexes emerged gradually across the northeast Asia, some of which spread eastward through northern Alaska and reached the previously uninhabited Greenland around 4500 years ago (11–13). Recent genetic studies have revealed complex and dynamic population histories in northeast Asia, shaped by multiple admixture and gene flow events. These studies reported substantial changes in human population genetic structure associated with migrations across the area (5, 14–17). Still, further investigation is required to fully assess the population dynamics of this vast geographical territory across a broad time span. Increased resolution can be achieved by analyzing ancient genetic data from unexplored regions and archaeological sites of northeast Asia. For example, genetic data from sites in the Trans-Baikal area carrying the earliest traces of post-LGM human occupation and from sites in Yakutia associated with the ancestors of the Paleo-Inuit Saqqaq cultural complex can provide new investigative venues. Here, focusing on these regions, we explore the population history of northeast Asia, particularly throughout the Lena, Angara, and Kolyma river basins and the Lake Baikal area. In studying a comprehensive sample of ancient genomes, we infer to what extent mobility, admixture processes, or even pandemics have affected northeast Asia over the Holocene. RESULTS AND DISCUSSION Genome-wide ancient DNA data We produced whole-genome sequence data from 40 ancient individuals spanning from the Late Upper Paleolithic to the Medieval era (table S1) and representing five distinct administrative regions in the Russian Federation encompassing Yakutia, Trans-Baikal, Cis-Baikal, Krasnoyarsk Krai, and Amur Oblast (Fig. 1). Genome sequence data from Yakutia include 10 individuals spanning a time period between c.16,900 and 2490 calibrated years before the present (cal BP) (0.03×–8.9× genome coverage); data from the Trans-Baikal area consist of 8 individuals dated to between c.8515 and 3000 cal BP (0.1×–4.7×); data from the Cis-Baikal area encompass 20 individuals spanning a time period between c.8980 and 550 cal BP (0.1×–14.5×); data from Krasnoyarsk Krai encompass an individual dated to c.4280 to 4085 cal BP (13.6×); and data from Amur Oblast cover an individual dated to c.1345 to 1270 cal BP (0.7×) (fig. S1 and table S1). Fifteen individuals were classified as biologically female, and 25 individuals were found to be male (table S1). All individuals were accredited to either Y macro-haplogroup Q or N and non-African mitochondrial macrohaplogroups of M, N, and R (18). We excluded biologically related individuals from the frequency-based analyses (table S1 and see Supplementary Text).  Fig. 1 Geographical and chronological information concerning the ancient individuals. (A) Geographical map showing the locations of the individuals sequenced in this study (orange, blue, and red). Genomes published elsewhere are shown as black (see table S2 for information about all published individuals used in comparative analysis). (B) Timeline showing the ages of the ancient individuals as calibrated years before the present. The degree of modern DNA contamination was negligible for all data (table S1). Principal components analysis (PCA) using present-day world populations (table S2) displayed a genetic gradient extending from west Eurasia to Central and East Asia (Fig. 2A). Projecting ancient individuals onto the space defined by the first two components indicated that the analyzed ancient northeast Asian individuals harbored genetic affinities to present-day populations of Central and East Asia (Fig. 2A). To infer post-LGM human population dynamics in northeast Asia, we first analyzed the oldest individual in our data and then investigated the population transformations in time until the Medieval era. |
|

