|
|
Post by Admin on May 27, 2020 19:31:37 GMT
Disentangling the action of positive and balancing selection on the Italian genomes
Genomic signatures ascribable to the action of positive and balancing selection on N_ITA and S_ITA ancestors were detected by computing the derived intra-allelic nucleotide diversity (DIND) and the number of segregating sites by length (nSL) scores, as well as by applying the BALancing selection LikElihood Test (BALLET). Genome-wide distributions obtained for these statistics were then used as input for gene network analyses aimed at testing the adaptive evolution of the Italian population groups under a model as close as possible to that of polygenic adaptation (see the “Methods” section). To focus on local adaptations that specifically characterize the ancestors of present-day Italians, we replicated these analyses on WGS data for the IBS and CEU populations, and we filtered out signatures of natural selection shared between them and the Italian clusters. In fact, although we cannot rule out the possibility that gene flow from other European and Mediterranean human groups has contributed to the evolution of these biological adaptations, this filtering approach enabled us to shortlist those selective signatures that are more plausibly ascribable to a combination of nature, duration, and intensity of the selective pressures that was peculiar of the Italian Peninsula.
According to the DIND test, RIMS2 and PCLO genes involved in insulin exocytosis (Additional file 1: Supplementary Results) were found to be subjected to positive selection in both N_ITA and S_ITA clusters (Additional file 1: Figure S5, Table S3). A gene network belonging to the Mucin type O-glycan biosynthesis pathway and formed by loci encoding for mucins, a family of glycosylated proteins that constitute the main protective barrier on mucosal surfaces and cellular membranes by preventing pathogens binding by steric hindrance [61], was instead supposed to have adaptively evolved only in S_ITA (Additional file 1: Figure S6, Table S3, Supplementary Results).
According to the nSL test, a gene network ascribable to the insulin secretion pathway, but made up of different loci with respect to those pointed out by DIND scores, characterized the N_ITA cluster (Additional file 1: Figure S5, Table S4). Among these genes, ADCY2, ADCY3, ADCY9, and GNAS are known to play a role in the regulation of lipolysis at the level of the adipose tissue, thermogenesis, and glucagon signaling (Additional file 1: Supplementary Results), with especially adenylate cyclase (ADCY) genes showing the largest number of connections in the network and participating to the longevity regulating pathway as well. Moreover, variants at two loci belonging to such a network and encoding for components of calcium voltage-gated channels (i.e., CACNA1C and CACNA1D) were previously reported to be involved in the development of type II diabetes (T2D) (Additional file 1: Supplementary Results). nSL results obtained for S_ITA corroborated those based on the DIND statistics as regards a gene network belonging to the mucin type O-glycan biosynthesis pathway (Additional file 1: Figure S6, Table S4) and further indicated genes from the basal cell carcinoma pathway as subjected to positive selection (Additional file 1: Figure S7, Table S4). Interestingly, most of these latter loci encode for frizzled G protein-coupled receptors (FZD) and Wnt glycoproteins that play a role in melanogenesis and participate in the mTOR signaling pathway as well (Additional file 1: Supplementary Results).
Events of balancing selection at two different gene networks were inferred for both the Italian population clusters (Additional file 1: Table S5). In detail, a network was found to be composed by several ADCY genes, some of which (i.e., ADCY2 and ADCY9) were the same putatively subjected also to positive selection in N_ITA, as well as by mitogen-activated protein kinase (MAPK) genes (i.e., MAPK1/ERK, MAPK8/JNK, and MAPK11/P38). These loci are known to play a role especially in the longevity regulating and FoxO signaling pathways (Additional file 1: Supplementary Results). The second network was instead made up of glycerol phosphate acyltransferase (GPAT/AGPAT/MBOAT), diacylglycerol kinase (DGKA), and lipid phosphatase (LPIN/PLPP) genes regulating the metabolism of glycerolipids, along with phospholipase (e.g., PLA2G/PLB) loci involved especially in the arachidonic acid metabolism (Additional file 1: Supplementary Results). Two gene networks were found to characterize exclusively the N_ITA group, being composed of aldehyde dehydrogenase (ALDH) genes important for glycolysis/gluconeogenesis and of protein kinase C (PRKC) and phospholipase (PLCG/PLCB) loci participating to the AGE-RAGE signaling in diabetic complications, glucagon signaling, and insulin resistance pathways (Additional file 1: Table S5). Finally, a further network turned out to be subjected to balancing selection in the S_ITA cluster, including PLA2G genes involved in the metabolism of arachidonic acid and MAPK1 and GRM1 loci that play a role in the FoxO signaling pathway (Additional file 1: Table S5, Supplementary Results).
Discussion
The clinal distribution of human genetic variation across Europe and the subtle divergence between groups from northern and southern regions of the continent are uniquely recapitulated at a micro-geographic scale by patterns of population structure observable along the Italian Peninsula [11,12,13, 16, 21]. To date, remarkable efforts have elucidated important aspects of the demography of the ancestors of modern Italians, which have contributed to their heterogeneous genetic background [14,15,16,17,18,19, 21, 22]. However, constraints imposed by the use of uniparental markers or of common autosomal SNPs affected the inferences drawn by these researches. Moreover, just a few studies attempted to complete the picture of the Italian genetic history with the investigation of local adaptations evolved by ancestral populations distributed along the peninsula in response to a wide range of environmental conditions [16, 23]. Additionally, none of them relied on data useful to test a model of polygenic adaptation mediated by natural selection slightly affecting many genes involved in the same biological function, but individually contributing a limited phenotypic effect, which has recently emerged as one of the predominant mechanisms of adaptive evolution of the human genome [35, 38].
In the attempt to overcome these issues, we aimed at depicting the demographic and adaptive history of the ancestors of present-day Italians by taking advantage of high-coverage WGS data. For this purpose, we first compared the examined genomes with genome-wide genotypes already available for the overall Italian population via a Procrustes analysis, demonstrating that they are representative of the two genetically homogeneous clusters (i.e., N_ITA and S_ITA) corresponding to the edges of the cline of Italian variation (Additional file 1: Figure S1). Moreover, fineSTRUCTURE clustering pointed to an appreciable divergence of these Italian groups (Fig. 1a), which is further supported by a low but highly significant genome-wide estimate of genetic differentiation (Fst = 0.0021; p value < 10−6). Previous studies have proven that, with the exception of Sardinians, N_ITA and S_ITA clusters encompass the most distinct ancestry components detectable at a considerable frequency in the Italian population and that, conversely, people from Central Italy present variable degree of admixture between them, but no additional private ancestry fractions [13, 15,16,17]. Therefore, the assembled WGS dataset enabled us to draw demographic and adaptive inferences according to a reliable approximation of the full spectrum of genetic components observable in the entire Italian gene pool.
|
|
|
|
Post by Admin on May 27, 2020 22:55:32 GMT
Late Glacial, Neolithic, and Bronze Age demographic processes left indelible signatures in the Italian genomes Our fineSTRUCTURE analysis further suggested that divergence between Italian clusters was reflected by a wide genetic Mediterranean “continuum” involving S_ITA and populations from Crete and the Caucasus as opposed to the affinity of N_ITA with groups from Continental Balkans (e.g., Bulgaria and Albania) (Fig. 1a). As for S_ITA, this peculiar pattern was recently proposed to be ascribable to Neolithic and Bronze Age contributions to the local gene pool originating from the Near East and the Caucasus. In particular, the Caucasus was identified as the potential source of a Bronze Age population movement that impacted Southern Italy approximately at the same time but independently from the well-known steppe-related migrations that occurred in Continental Europe. Clear marks of the latter demographic process were instead observed in Northern Italy, as well as in Central and Northern Balkans [17]. The results from the analysis of residuals calculated by contrasting N_ITA and S_ITA outgroup f3 statistics and using a large panel of aDNA samples are consistent with the hypothesis mentioned above. In fact, increased shared genetic ancestry with Chalcolithic/Bronze Age and, especially, Neolithic remains from Anatolia, Armenia, Near East, and Greece was inferred for S_ITA with respect to N_ITA, with the largest residuals pointing to the relationships of S_ITA with populations from Iran and the Levant dating back to the Neolithic (Fig. 2b). These findings confirm the early positioning of Southern Italy at one of the westernmost edges of the extensive Mediterranean corridor that mediated the diffusion of farming from Southeastern Europe [5, 28, 79] and suggested Neolithic processes having left some of the most substantial traces (e.g., in terms of Anatolian-Neolithic-related and Caucasus hunter-gatherer ancestries) in the genetic background of S_ITA people. Moreover, they suggested that subsequent Chalcolithic/Bronze Age population movements having influenced the S_ITA gene pool have plausibly originated from Southern Caucasus and Anatolia and reached the Italian Peninsula through a Mediterranean route [7]. In addition to gene flow that occurred during historic times along the same path (Fig. 1c, Additional file 1: Tables S1-S2), this ancient connection contributes to explain also the patterns of haplotype sharing with present-day populations from the Near East and Southern Caucasus that were observed predominantly for S_ITA (Fig. 1b). On the contrary, a more substantial ancestry shared by N_ITA with Western European remains dated to the Copper Age or associated with the Bell Baker complex was observed along with their increased affinity to the Central and Eastern European Bronze Age samples. Again, this is concordant with N_ITA chromosome painting profiles and ancestry proportions shared with modern groups such as the Basques and Eastern/Northern Europeans (Fig. 1b, c; Additional file 1: Table S1). Interestingly, signatures ascribable to relationships with considerably more ancient groups, including Eastern, Western, and Scandinavian hunter-gatherers and samples belonging to the Late Glacial “El Miron Cluster” also emerged, with the largest outgroup f3 residuals being associated with hunter-gatherer specimens from the Balkans, Latvia, and Switzerland, as well as with the post-Ice Age “Villabruna cluster” (Fig. 2a). Contrarily to what is supposed for Sardinians [27], we speculate that this pattern is only partially ascribable to a direct link of present-day N_ITA with local Upper Paleolithic groups. Instead, this is in line with the hypothesis that population movements that involved the Italian Peninsula during and after the Neolithic have replaced a great part of local Paleolithic genetic backgrounds [13, 16, 17]. Accordingly, the observed affinity with hunter-gatherer samples might be likely due to the resurgence of genetic components proper of the early European founder population because of demographic processes that occurred during the Late Glacial and, particularly, the Bronze Age. This is suggested by N_ITA affinity with “El Miron Cluster,” which is dated to around 19–14 kya and was found to attest a post-Ice Age re-expansion from southwestern European refugia of an ancestry fraction that was widespread all over Europe between 34 and 26 kya [4]. The close relationship with the “Villabruna Cluster” might instead reflect the impact that the diffusion of the Epigravettian culture exerted on the ancestral N_ITA gene pool since the end of the LGM [4]. Finally, groups migrated from the Eurasian Steppe during the Early Bronze Age, such as Yamnaya pastoralists, have been previously demonstrated to present substantial pre-Neolithic ancestry fractions in addition to their peculiar steppe-related genetic component [2, 3, 80] Consequently, these population movements are supposed to have contributed to raise again the Eastern hunter-gatherer ancestry in Western Europeans since around 4.5 kya, as testified by several remains belonging to the Bell Baker complex and including the Iberian ones that showed increased shared genetic drift with N_ITA [8]. Overall, the distinct ancestry composition described for N_ITA and S_ITA clusters fits well also with the demographic scenario depicted by modeling their ancient and recent population history with the coalescent-based SMC++ method (Fig. 3). The seemingly higher Ne inferred for S_ITA with respect to N_ITA until the beginning of the Late Glacial might be compatible with the hypothesis of a refugee role played by Southern Italy during the LGM (see also the paragraph below about climate-mediated adaptations) [14, 22, 28]. However, it is not possible to evaluate the actual statistical significance of this subtle Ne difference, at least as concerns the period that predates the inferred population split time. Moreover, this pattern might be also ascribable to the more substantial level of gene flow from diverse populations experienced by S_ITA with respect to N_ITA, as proposed to explain the differences in Ne observed between Southern and Continental European groups [78]. More interestingly, appreciable genetic differentiation between N_ITA and S_ITA can be approximately dated back to just after the end of the LGM (Fig. 3), if we consider that the obtained population split time (i.e., 9 kya) represents a rough underestimate due to a clear violation of the assumption of negligible post-divergence gene flow between clusters made by the SMC++ model. This is thus in line with a scenario assuming that the Late Glacial demographic processes described above have represented the first step in the cascade of events that differentially shaped the gene pool of present-day N_ITA and S_ITA groups. Climate-mediated adaptive evolution at insulin-related genes especially in Northern Italy Both selection scans performed to test for the occurrence of positive and balancing selection suggested a complex pattern of adaptive evolution at insulin-related genes in the Italian people. In detail, selective events able to modulate insulin exocytosis from pancreatic beta cells were supposed to have occurred in the common ancestors of N_ITA and S_ITA clusters (Additional file 1: Figure S5, Table S3, Supplementary Results). Events of positive selection presumably more recent were instead found to characterize exclusively people from N_ITA, being distributed among ten genes that play a role at different levels of the signaling cascade leading to insulin secretion and that regulate key processes contributing to glucose homeostasis (Additional file 1: Figure S5, Table S4, Supplementary Results). Interestingly, the most pervasive signature of selection was observed at ADCY genes (especially ADCY3), which are fundamental for controlling thermogenesis [45] and adiposity [46, 47] and have been proven to modulate susceptibility to T2D and obesity (Additional file 1: Supplementary Results). In line with these findings, analyses testing for balancing selection pointed to adaptive events specific of the N_ITA cluster and mediated by ALDH genes involved in glycolysis and gluconeogenesis or by PRKC and PLCG/PLCB loci playing a role in pathological mechanisms underlying insulin resistance and the onset of diabetic complications (Additional file 1: Table S5). According to this body of evidence, we can speculate that climate- and tightly linked dietary-related selective pressures have presumably played a role in determining the described selection signatures (Fig. 4). The few ones shared between N_ITA and S_ITA clusters might indeed represent a legacy ascribable to the retreating of human groups distributed along the peninsula towards Central/Southern Italian refuge areas during the LGM [14, 22, 28]. There, northern and southern ancestral populations likely admixed and lived in forest-steppe habitats for around 10,000 years. This period was long enough to have possibly triggered optimization of energy metabolism in response to a cold environment in which animal-based high-energy/high-fat diets represented the main nutritional resource, as testified by isotope analyses on archeological records ascribable to the Gravettian and Epigravettian cultures [81]. This hypothesis is in agreement with evidence pointing to most of the adaptive events inferred so far for populations of Western European ancestry being dated to the LGM and correlating with environmental variables that suggest climate cooling and short-term temperature instability as some of the main selective pressures [82, 83]. Fig. 4 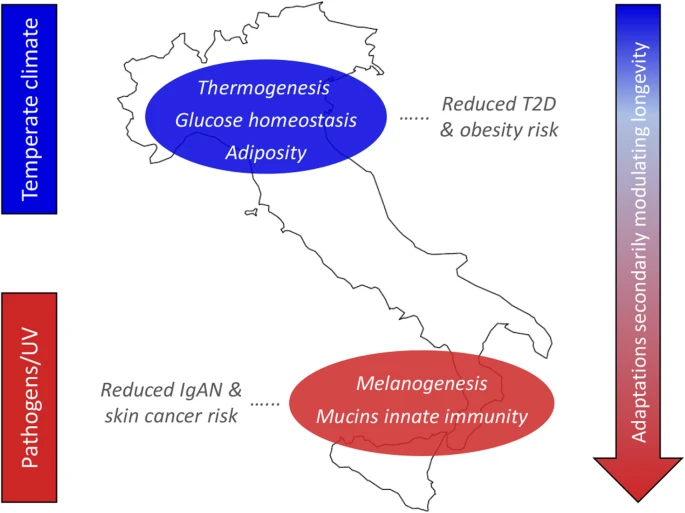 Adaptive events evolved by ancestors of N_ITA/S_ITA clusters and their health implications for present-day Italians. The putative selective pressures having plausibly prompted local adaptations are displayed on the left, while biological processes subjected to natural selection are reported on the map along with their impact on present-day disease susceptibility. Distribution of biological adaptations having the potential to modulate the longevity phenotype (e.g., involving the mTOR signaling, arachidonic acid metabolism, and FoxO signaling pathways) in the overall Italian population, but especially in people from Southern Italy, is represented by the arrow on the right. Putative selective pressures, biological processes, and distribution of adaptations potentially modulating longevity are color-coded as follows: N_ITA, blue; S_ITA, red With progressive climate warming during the Late Glacial, some groups moved back from refuge areas to repopulate the Northern Italian regions and, differently from populations expanding southwards who soon experienced again a Mediterranean climate, they continued to be subjected to selective pressures similar, although less extreme, to those acting during the LGM. For several other millennia, these people had to cope with a temperate climate characterized by cold winter seasons and have been more affected than Southern Italian groups by the climate changes that occurred in Continental Europe since the Bronze Age until recent historical times [84,85,86]. Although we cannot rule out the possibility that recent and differential gene flow from populations exposed to diverse environmental conditions contributed to exacerbate the differentiation of selection signatures observed between N_ITA and S_ITA groups, the climate picture described above has the potential to have represented a non-negligible factor in the evolution of more pervasive selective events by the ancestors of N_ITA, which extend beyond the simple regulation of insulin secretion to biological pathways able to modulate cell sensitivity to it, along with the metabolism of the adipose tissue and the expression of genes promoting thermogenesis (Fig. 4). This adaptive scenario fits well also with the picture of early differentiation between N_ITA and S_ITA clusters revealed by the SMC++ analysis, which become appreciable just since a few thousand years after the end of the LGM (Fig. 3). Interestingly, having targeted genes whose dysfunction is known to play a role in the development of T2D and/or obesity, most of the inferred N_ITA-specific signatures of positive and balancing selection seem to be ascribable to evolutionary events with potential biomedical relevance. For instance, adaptive evolution at these loci might have contributed to make people from Northern Italy less prone to develop such diseases even in the challenging nutritional environment imposed by modern lifestyles (Fig. 4). This is in line with the values of T2D incidence almost reduced by half in N_ITA with respect to S_ITA [87] and may further support recent attention drawn by our best candidate gene (i.e., ADCY3) as a promising target for the development of anti-obesity drugs [88]. |
|
|
|
Post by Admin on May 28, 2020 0:41:15 GMT
Pathogens and solar radiation may have triggered adaptations peculiar to Southern Italy
When considering adaptive events specific to S_ITA, genes encoding for mucins that prevent pathogens binding at the level of mucosal surfaces and loci participating in melanogenesis emerged as putative targets of positive selection (Fig. 4, Additional file 1: Figure S6-S7, Tables S3-S4).
Among mucin genes, C1GALT1 represented the central node of the two identified gene networks (Additional file 1: Supplementary Results), and several genome-wide association studies previously reported a correlation of some of its variants to immunoglobulin-A nephropathy (IgAN), which is the most common human kidney inflammation [57]. Interestingly, epidemiologic data highlighted a considerably higher IgAN prevalence in Northern Italian regions than in Southern Italy [58]. According to this picture, and because several microorganisms are known to have evolved chemical strategies aimed at enzymatically inactivating mucins to elude mucosal/cellular barriers [57], we can hypothesize that some adaptive events that possibly occurred in response to these pathogens may have contributed to reduced S_ITA susceptibility to IgAN (Fig. 4). Among microorganisms able to inactivate mucins, Pseudomonas aeruginosa, the parasitic amoebozoan Entamoeba histolytica, and the proteobacterium Burkholderia cepacia present a geographical distribution that correlates negatively to that of IgAN and positively to environmental temperature (Additional file 1: Supplementary Results). Therefore, we can speculate that infections by these pathogens or by closely related species might have been more frequent in the past in Southern Italian regions than in northern ones, having potentially represented selective pressures able to trigger adaptive evolution of mucin genes in the ancestors of S_ITA.
Environmental conditions characterized by a mean value of annual solar radiation nearly double with respect to Northern Italy [89] might have played a role in the evolution of S_ITA-specific selection signatures at FZD/Wnt genes that are involved in melanogenesis (Fig. 4). In fact, being responsible for basal and ultraviolet (UV)-induced melanin production, melanocytes expressing these genes represent a frontline defense against harmful UV-B radiation. FZD genes found to have adaptively evolved in S_ITA act as receptors of Wnt protein ligands that showed comparable selection signatures and regulate the expression of the microphthalmia-associated transcription factor (MITF) [90]. By controlling pigmentation genes (e.g., TYR, TYRP1, and TYRP2), MITF is the main modulator of melanogenesis in response to environmental stimuli and was also proposed to exert an oncogenic role in several skin cancers [91]. This might explain the involvement of the identified FZD/Wnt genes under selection in the basal cell carcinoma pathway. Overall, these selective events could have mediated adaptations of S_ITA ancestors aimed at preventing skin micronutrient photodegradation and/or impairment of sweat gland-mediated thermoregulation due to UV damage [92]. Because substantial UV exposure represents the main risk factor for developing basal cell carcinoma and other types of skin malignancies, these adaptive mechanisms might have also indirectly contributed to reduce the predisposition of modern S_ITA to such diseases (Fig. 4). This hypothesis seems to be in agreement with the almost halved incidence of melanomas reported for Southern Italian regions with respect to northern ones [93].
Pleiotropic adaptive events potentially modulating longevity in the Italian population
Several selection signatures observed for the overall Italian population, but resulting more pronounced in S_ITA, pointed to adaptive events mediated by biological processes that are known to play a role also in the achievement of the longevity phenotype (Fig. 4, Additional file 1: Table S5). Interestingly, this is in line with recent findings showing that Italian centenarians genetically cluster with people from Central/Southern Italy regardless of their micro-geographic origins [76]. Among the most relevant signatures, we emphasize S_ITA-specific positive selection at FZD/Wnt genes that take part in the mTOR signaling pathway as well. Overall, variants at loci belonging to this pathway have been demonstrated to be able to delay age-related diseases and/or to directly influence longevity even in the human species [67] (Additional file 1: Supplementary Results).
Identification of footprints of balancing selection at genes involved in the metabolism of arachidonic acid complements previous findings obtained for the general Italian population and for centenarians from the peninsula [16, 76] (Additional file 1: Supplementary Results). The emerging picture suggests that these adaptive events may have evolved in response to specific pathogens and secondarily maintained in the Italian gene pool alleles useful to contrast the side effects of modern pro-inflammatory diets, thus contributing to longevity [76]. Balancing selection was found to have targeted also several genes involved in the FoxO signaling, which provides monitoring of stress stimuli, such as dietary restriction, absence of insulin or insulin-like growth factors, and uptake of intracellular pathogens, being associated with exceptional longevity as well [71] (Additional file 1: Supplementary Results). Accordingly, both nutritional and pathogen-related selective pressures might have triggered such adaptive events, which have been observed so far only at the single-gene level for FOXO3 [94].
Conclusions
By taking advantage from high-coverage WGS data, the present study has had the opportunity to infer the demographic and adaptive history of the ancestors of modern Italians with an unprecedented level of resolution. In particular, we provided new evidence for early differentiation dating back to the Late Glacial between population clusters that represent the edges of the cline of Italian variation, as well as for Neolithic and distinct (i.e., steppe-related versus Anatolian/Mediterranean) Bronze Age demographic processes having then continued to differentially shape the gene pool of groups distributed along the peninsula. Moreover, we proposed climate-related selective pressures as potential factors having influenced adaptive evolution at insulin-related genes especially in the ancestors of Northern Italians. By regulating glucose homeostasis, adiposity, and thermogenesis in response to high-calorie diets adopted to cope with energetically demanding environmental conditions, these adaptive events might have also contributed to make people from Northern Italy less prone to develop T2D and obesity despite the challenging nutritional context imposed by modern lifestyles. Conversely, possible adaptations against pathogens and modulation of melanogenesis in response to high UV radiation are supposed to have played a role in reduced susceptibility of people from Southern Italy respectively to immunoglobulin-A nephropathy and skin cancers. Finally, multiple adaptive processes evolved by the overall Italian population, but having resulted more pronounced in people from the southern regions of the peninsula, were found to have the potential to secondarily modulate the longevity phenotype. Therefore, by pinpointing genetic determinants underlying biological adaptation of Italian population clusters in response to locally diverging environmental contexts, the present study succeeded in disclosing also valuable biomedical implications of such evolutionary events. Coupled with the identification of the demographic processes having predominantly shaped the present-day heterogeneous Italian genomic background, this supports once again the usefulness of an evolutionary approach in the dissection of the deep causes of human populations’ health and disease, and highlighted important dynamics that contributed to the formation of the Continental and Southern European genomic landscapes.
|
|
|
|
Post by Admin on Apr 24, 2021 20:05:53 GMT
Early Alpine occupation backdates westward human migration in Late Glacial Europe Summary Before the end of the Last Glacial Maximum (LGM, ∼16.5 ka ago)1 set in motion major shifts in human culture and population structure,2 a consistent change in lithic technology, material culture, settlement pattern, and adaptive strategies is recorded in Southern Europe at ∼18–17 ka ago. In this time frame, the landscape of Northeastern Italy changed considerably, and the retreat of glaciers allowed hunter-gatherers to gradually recolonize the Alps.3, 4, 5, 6 Change within this renewed cultural frame (i.e., during the Late Epigravettian phase) is currently associated with migrations favored by warmer climate linked to the Bølling-Allerød onset (14.7 ka ago),7, 8, 9, 10, 11 which replaced earlier genetic lineages with ancestry found in an individual who lived ∼14 ka ago at Riparo Villabruna, Italy, and shared among different contexts (Villabruna Cluster).9 Nevertheless, these dynamics and their chronology are still far from being disentangled due to fragmentary evidence for long-distance interactions across Europe.12 Here, we generate new genomic data from a human mandible uncovered at Riparo Tagliente (Veneto, Italy), which we directly dated to 16,980–16,510 cal BP (2σ). This individual, affected by focal osseous dysplasia, is genetically affine to the Villabruna Cluster. Our results therefore backdate by at least 3 ka the diffusion in Southern Europe of a genetic component linked to Balkan/Anatolian refugia, previously believed to have spread during the later Bølling/Allerød event. In light of the new genetic evidence, this population replacement chronologically coincides with the very emergence of major cultural transitions in Southern and Western Europe. Results and discussion Riparo Tagliente represents the earliest available evidence of human occupation of the southern Alpine slope5 while the major glaciers in the area started withdrawing at 17.7–17.3 cal BP (2σ)6,13 and is therefore critical to address questions on the impact of human movement in this time frame (Figure 1; STAR Methods). We performed anthropological (STAR Methods, Method details) and genetic analyses to assess the biological background of the sampled individual. The hemimandible, which is affected by focal cemento-osseous dysplasia (Figures 2 and S1; STAR Methods, Method details), was also directly dated to independently ascertain its chronology and the possible contemporaneity with contextual, post-cranial human remains from a partially preserved burial (Tagliente1; STAR Methods).1415 The root of the first molar (LM1) from the hemimandible of Tagliente2 was directly dated to 16,980–16,510 cal BP (95.4% probability using IntCal2016 in OxCal v.4.3;17 Data S1A), confirming the attribution to the Late Epigravettian chronological range, i.e., the same cultural context as Tagliente1 (16,130–15,560 cal BP; 2σ range obtained using Reimer et al.;16 Data S1A; STAR Methods, Method details).  Figure 1 Geographical, ecological, and cultural context of the study (A) Palaeogeographic map of Europe during Late Glacial, centered at 17 ka ago (see Method details). Colored areas refer to the distribution of Epigravettian and Magdalenian material culture at 17 ka ago, although white symbols indicate the geographic location of the main sampling sites discussed in the text (~30–8 ka ago). (B) 3D lateral view of the hemimandible Tagliente2 with roots, pulp chambers, dentine, and enamel of the preserved teeth, as well as the cementome (in red) between the distal side of P3 root and the mesial root side of M1 (Figure S1). (C) Comparison between palaeoclimate, palaeoenvironmental, and cultural proxies over the 30–11 ka cal BP time span. Key to panels is as follows: (1) reconstruction of Southern Alpine past vegetation;3,6,18,19,20 (2) Eurasian major megafaunal transitions (regionwide extirpations or global extinctions, or invasions, of species or major clades);21,22 (3) NGRIP δ18O record in 20 years means on the GICC05 timescale;23 and (4) material cultural sequence for Eastern and Southern Europe. Chronology for Tagliente2 (2σ obtained using Reimer et al.16; Data S1A) is marked in blue. |
|
|
|
Post by Admin on Apr 24, 2021 21:42:05 GMT
 Figure 2 Tagliente2 virtual (left) and physical (right) section On the left, microCT distal view of the premolar and its pathological cementum tissue. On the right, histological section is shown. Magnification (250×) of the cementum tissue colored by hematoxylin/eosin. B, buccal; L, lingual; scale bar represents 0.5 mm (see Figure S1, Data S1B, and STAR Methods). We extracted DNA from five samples taken from mandibular and tooth tissues and screened for the presence of endogenous human DNA through a pooled whole-genome sequencing. One of the healthy mandibular samples yielded sufficient endogenous DNA (5.06%) and was re-sequenced to achieve a total genome-wide coverage of 0.28×, yielding 266K SNPs overlapping with the 1240K Human Origins SNP Array. We also provide a number of missense variants found to overlap with genes known to be involved with cementoma insurgence, which, given low coverage and ancient DNA degradation, are reported with no further interpretation (Data S1B; STAR Methods, Method details). Overall contamination estimated from mtDNA was 2.158% and 0.60%–1.53% from the X chromosome (Data S1C). The mtDNA haplogroup is a basal U2′3′4’7’8’9 (Data S1C and S1D), consistent with a European Palaeolithic individual (Figure 3A), also shared by Rigney1 (15.5 ky cal BP)9 and Paglicci Accesso Sala 2 Rim P (∼13 ka ago).24 X/autosome coverage ratio in the order of 0.56 suggests the individual was male, in accordance with results of morphological analysis (STAR Methods, Method details), and the chromosome Y haplogroup was estimated to be I2, which captures the majority of diversity in Europe after ∼14 ka ago (Figure 3B; Data S1E). Most samples dated to this period fall within a single mtDNA (U5b)10 and chrY (I2)9 branch, flagging a putative expansion from a single founding population.  Figure 3 Uniparental haplogroups of Tagliente2 (A) mtDNA haplogroup of Tagliente2 (in gold; Data S1C and S1D; Figure S4) within a number of pre-(green) and post (blue)-Villabruna samples. (B) chrY haplogroup of Tagliente2 (gold; Data S1C and S1E) surrounded by post-Villabruna samples (including Bichon [BC]). Y haplogroup splits are drawn according to the dater estimates based on high-coverage modern sequences.25 The ancient individuals are mapped on this tree, considering the available haplogroup-informative available SNP data, and private mutations in the ancient samples have been ignored. |
|

