|
|
Post by Admin on Oct 2, 2019 21:23:01 GMT
An international team of researchers has analyzed remains from ten archaeological sites in England, France, Germany, Russia, and Switzerland to gain insight into the different stages of the second plague pandemic (14th-18th centuries) and the genetic diversity of Yersinia pestis during and after the Black Death. In a study published in Nature Communications, the researchers reconstructed 34 Y. pestis genomes, tracing the genetic history of the bacterium, which revealed key insights into the initiation and progression of the second plague pandemic in Europe. 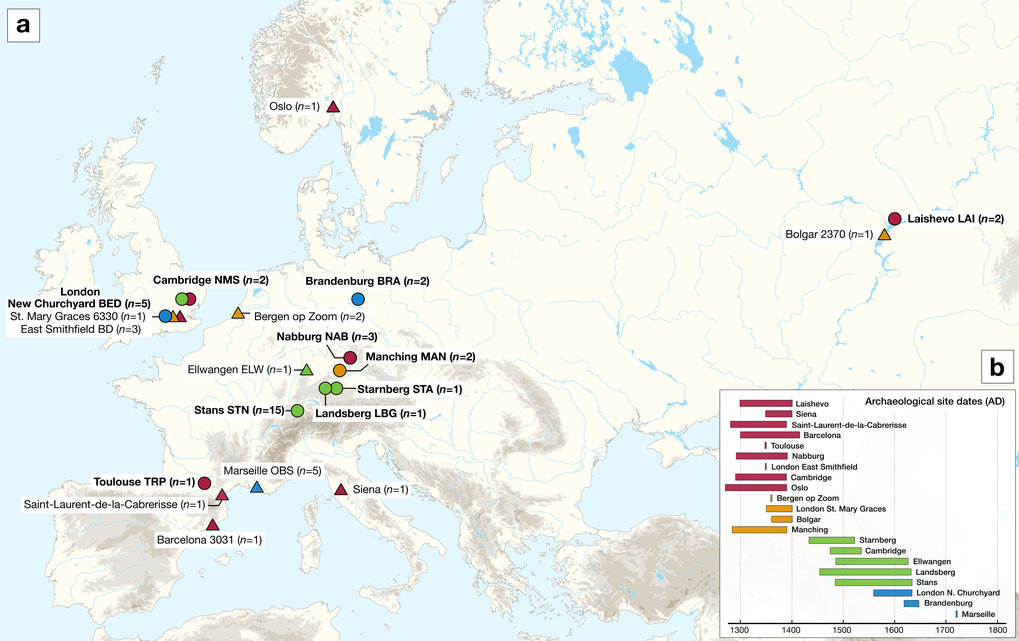 The second plague pandemic, which began with the Black Death in the mid-14th century and continued with devastating outbreaks in Europe and the vicinity until the 18th century, decimated the continent, causing the death of up to 60% of the population. But where did this strain of Yersinia pestis, the plague causing bacterium, come from? And how did it evolve and expand once it arrived? A likely point of entry for Y. pestis during the second pandemic Despite the ubiquity of the Black Death in historical texts and the popular imagination, the entry point of the Y. pestis bacterium at this time and the route it traveled through Europe remain unclear, due to a lack of data from early outbreaks and a general scarcity of published ancient Y. pestis genomes. In the current study, researchers reconstructed plague genomes from the teeth of 34 individuals, including two from Laishevo, in the Volga region of Russia, and found a single strain that is ancestral to all second pandemic strains. In addition, the researchers observe an absence of genomic diversity from samples during the Black Death. “These findings indicate a single entry of Y. pestis into Europe through the east”, explains first author Maria Spyrou of the Max Planck Institute for the Science of Human History. “However, it is possible that additional interpretations may be revealed with future discoveries of un-sampled diversity in western Eurasia”, she notes.  Persistence of Y. pestis within Europe Although the researchers found that the European-wide Black Death was likely caused by a single strain, analysis of genomes from later in the pandemic shows the emergence of a lineage displaying a higher genetic diversity. “In the later phase of the second pandemic, we see the development of multiple branches within Europe, which suggests that plague was maintained in different local foci”, says Marcel Keller, co-first author of the Max Planck Institute for the Science of Human History. “No modern descendants of this lineage have been found to date, possibly indicating the extinction of these reservoirs.” The researchers also identified a deletion including two virulence-related genes from genomes within this second lineage. Interestingly, genomes from the late stages of the first plague pandemic have shown a deletion in the same region. “Given that this deletion occurred in lineages from the first and second pandemic, both now extinct, determining how these genes impact maintenance in human and flea hosts would be an important area for future study”, comments Kirsten Bos, research group leader of the Max Planck Institute for the Science of Human History.  The current study provides new perspectives into the initiation and progression of the second plague pandemic and adds significantly to the database of published ancient Y. pestis genomes. “We have shown that extensive analysis of ancient Y. pestis genomes can provide unique insights into the microevolution of a pathogen over a period of several hundred years”, says senior author Johannes Krause, Director of the Department of Archaeogenetics at the Max Planck Institute for the Science of Human History. In the future, integrating this data into disease modelling efforts, in conjunction with data from other areas such as climate science, epidemiology and history, will be important for better understanding the second plague pandemic. Abstract The second plague pandemic, caused by Yersinia pestis, devastated Europe and the nearby regions between the 14th and 18th centuries AD. Here we analyse human remains from ten European archaeological sites spanning this period and reconstruct 34 ancient Y. pestis genomes. Our data support an initial entry of the bacterium through eastern Europe, the absence of genetic diversity during the Black Death, and low within-outbreak diversity thereafter. Analysis of post-Black Death genomes shows the diversification of a Y. pestis lineage into multiple genetically distinct clades that may have given rise to more than one disease reservoir in, or close to, Europe. In addition, we show the loss of a genomic region that includes virulence-related genes in strains associated with late stages of the pandemic. The deletion was also identified in genomes connected with the first plague pandemic (541–750 AD), suggesting a comparable evolutionary trajectory of Y. pestis during both events. |
|
|
|
Post by Admin on Oct 3, 2019 18:40:47 GMT
One of the most devastating pandemics of human history was the second plague pandemic, which began with the infamous Black Death (BD, 1346–1353 AD) and continued with recurrent outbreaks in Europe, the Near East and North Africa until the 18th century AD1,2. Its causative agent, Yersinia pestis3, is a highly virulent bacterium that causes bubonic, pneumonic, and septicaemic plague and today is maintained among wild rodent populations in eastern Europe, Asia, Africa and the Americas4,5,6. The first historically documented outbreaks of the second pandemic seem to have occurred in 1346 in the Lower Volga and Black Sea regions1,7. Subsequently, the bacterium dispersed through the rest of Europe over the next seven years, causing reductions in the human population estimated to be as high as 60%1. Recent studies on ancient Y. pestis DNA from medieval plague victims have contributed insights into these initial stages of the pandemic. Specifically, mid-14th-century Y. pestis genomes reconstructed from Saint-Laurent-de-la-Cabrerisse (southern France)8, Barcelona (Spain)9, London (England)10 and Oslo (Norway)8 were shown to be identical, suggesting the rapid dispersal of a single strain across Europe during the BD. Recently, the analysis of an additional low-coverage genome from Siena, Italy (BSS31)8, revealed the purported existence of Y. pestis strain diversity during the BD, a possibility that should be further explored. After the BD, plague was a common scourge in Europe as evidenced by the thousands of recorded epidemics it supposedly caused between 1353 and the late 18th century2,11. Whether these were caused by multiple introductions of the disease from an Asian source or by its local persistence in Europe is currently a topic of debate9,12,13,14. While data from climatic proxies are considered as supportive of the former hypothesis12, genetic evidence is interpreted in both directions8,9,13. To date, ancient Y. pestis genomes from epidemics closely succeeding the BD in Europe have been sequenced from late-14th-century individuals in Bergen op Zoom (Netherlands), London (England) and the Middle Volga region of Russia. They cluster on a phylogenetic lineage that is a precursor to strains associated with the 19th-century third plague pandemic9,15,16, and thus provide a link between medieval and modern plague. Moreover, Y. pestis genomes recovered from Ellwangen, Germany (1485–1627 calAD), and the Great Plague of Marseille in France (L’Observance, 1720–1722 AD) cluster on an independent lineage, here termed the “post-BD” lineage, that is to date unidentified among modern Y. pestis diversity. Both lineages descended from the strain associated with the BD and, hence, likely represent plague’s legacy in or around Europe after 1353. At present, the source of the second pandemic and the route that the bacterium followed during its course of entry into Europe remain hypothetical since genomic data from early outbreaks in western Russia have thus far been elusive. In addition, the limited number of published ancient Y. pestis genomes9,10,14 challenges our ability to construct hypotheses regarding the number of lineages responsible for the numerous post-BD outbreaks in Europe2,11 and whether they derived from a single or multiple disease reservoirs. Here, we take steps to overcome these limitations by expanding the number of available Y. pestis genomes from multiple time periods and locations in order to gain additional knowledge on the early stages of the second pandemic, and to study the genetic diversity of the bacterium present in Europe after the BD. Additionally, we present a reanalysis of recently published data from the same time period8. Our results support the entrance of Y. pestis into Europe through the east during the initial wave of the pandemic and consistently demonstrate an absence of genetic diversity in the bacterium during the BD. Moreover, our genomic analysis of post-BD outbreaks from central and western Europe suggests the local diversification of an extinct Y. pestis lineage between the late-14th and 18th centuries that may have resided in more than one disease reservoir. Results Sample screening for signatures of Y. pestis DNA Two approaches were used for the assessment of Y. pestis DNA in tooth specimens (n = 206) from ten archaeological sites spanning the 14th–17th centuries AD in Europe (Fig. 1, Supplementary Figs. 1–10 and Supplementary Note 1). First, a qPCR screening approach was employed for detection of the Y. pestis-specific gene, pla, located on the pPCP1 plasmid17 in 180 specimens from the cities of London (n = 40) in England, Toulouse (n = 42) in France, Brandenburg an der Havel13 (n = 3), Landsberg am Lech (n = 10), Manching-Pichl13 (n = 28), Nabburg (n = 12) and Starnberg (n = 3) in Germany, Laishevo (n = 10) in the Volga region of Russia, and Stans (n = 32) in Switzerland. Extracts from 41 teeth across these sites tested positive for pla (Supplementary Table 1). All extraction negative controls were free of amplification products. Amplification products from putatively positive individuals were not sequenced, as the presence of Y. pestis was subsequently assessed through whole-genome capture and high-throughput Illumina sequencing. 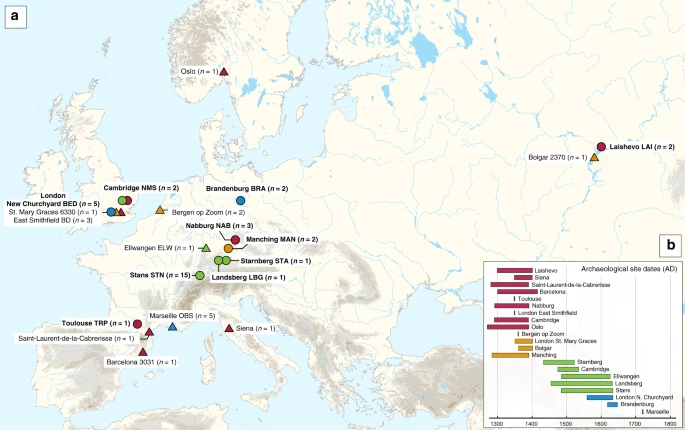 Fig. 1 In addition, shotgun Next Generation Sequencing (NGS) data from individuals unearthed at the New Museums site (Augustinian Friary) in Cambridge (n = 26) were screened for Y. pestis with the MEGAN alignment tool (MALT)18 (see Methods). The output was post-processed within the pathogen screening pipeline HOPS19. The assessment of shotgun NGS reads produced from non-uracil-DNA-glycosylase (non-UDG) libraries revealed the potential presence of Y. pestis DNA in four individuals (Supplementary Table 2, Supplementary Fig. 11). Y. pestis in-solution capture and whole-genome reconstruction We prepared UDG-treated libraries20,21 from all putatively positive samples and used a Y. pestis whole-genome in-solution capture approach22 combined with high-throughput sequencing for the retrieval of 1,299,105–79,055,317 raw reads per sequenced library. All data were mapped against the Y. pestis CO92 reference genome (NC_003143.1)3. This resulted in 86,278–3,822,030 unique mapping reads yielding 1.1–80.1-fold coverage across 34 individuals that span the time transect between the 14th and 17th centuries in Europe (Supplementary Table 3). More specifically, we could retrieve two Y. pestis genomes from Cambridge (England), five from London (England), one from Toulouse (France), three from Nabburg, two from Manching-Pichl13, one from Starnberg, one from Landsberg am Lech, two from Brandenburg an der Havel13 (all from Germany), two from Laishevo (Russia) and 15 from Stans (Switzerland). Of those, 24 isolates showed at least 50% of the reference genome covered at 5-fold (Table 1), which allowed for their confident inclusion in phylogenetic analysis. In addition, we nearly tripled the genomic coverage of the published “549_O” isolate from Ellwangen, Germany (now reaching 14.1-fold), which was previously processed by array-based capture using a different probe design9 (Supplementary Table 3). |
|
|
|
Post by Admin on Oct 4, 2019 18:23:35 GMT
Y. pestis phylogenetic reconstruction To infer genetic relationships between the new and previously published Y. pestis isolates, we constructed phylogenies using the maximum likelihood (ML) method, allowing for up to 3% missing data (97% partial deletion) to accommodate lower coverage genomes. As a reference dataset, we used 233 modern isolates23,24,25,26,27 (as listed in ref. 28), which represent most of the published Y. pestis genetic diversity. In addition, we included previously published second pandemic isolates (n = 15)8,9,10,14, a 6th-century AD isolate from Germany29, a 2nd- to 3rd-century AD isolate from the Tian Shan mountains in Kyrgyzstan30, as well as three Bronze Age isolates from the Altai and Volga regions31,32 (Supplementary Fig. 12). All newly reconstructed genomes appear on Branch 1 and are closely related to the previously published second pandemic isolates from Europe (Fig. 2), thus confirming their authenticity. In addition, they seem to represent a diverse group of strains that were present across Europe between the 14th and 18th century AD (Fig. 2, Supplementary Data 1). A number of genomes (NAB005, BRA003, STN011 and STN004) were excluded from further analyses as they showed evidence of excess heterozygosity, which is atypical of bacterial genomes (Supplementary Fig. 13). This phenomenon likely arises from enrichment of non-target DNA stemming from closely related organisms, an issue frequently encountered in ancient metagenomic datasets18,29,33. Moreover, these genomes had notably longer branch lengths in comparison to other contemporaneous isolates from the same archaeological contexts (Supplementary Fig. 14). Their assessment using the recently developed SNPEvaluation tool28 (see Methods) classified their private SNP calls as false-positive, suggesting that the observed branch lengths are erroneous (Supplementary Data 2). Similarly, the previously published SLC1006 and BSS31 genomes8 were also excluded from further analyses as they also showed high heterozygosity (Supplementary Fig. 15) and exceedingly longer branch lengths compared to other 14th-century Y. pestis genomes (Supplementary Figs. 14 and 16). 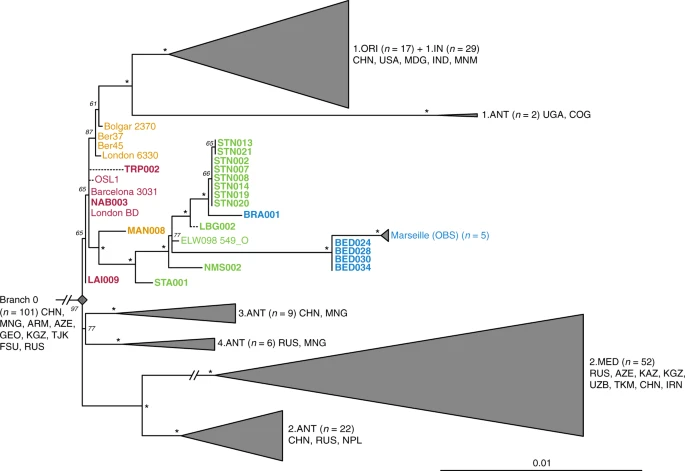 Fig. 2 Our phylogenetic reconstruction shows that the LAI009 isolate from Laishevo is ancestral to the BD isolates from southern, central, western and northern Europe, as well as to the previously published late 14th-century isolates from London (6330)10 and Bolgar City9 (Fig. 2). This genome possesses only one derived SNP distinguishing it from the N07 polytomy that gave rise to Branches 1–4 (Fig. 2; Supplementary Data 1)23. Since all other second pandemic genomes share an additional derived SNP on Branch 1, we interpret LAI009 as the most ancestral form of the strain that entered Europe during the initial wave of the second pandemic that has been identified to date. Regarding the central and western European genomes, NAB003 from Nabburg does not show differences compared to previously published BD genomes from London and Barcelona9,10. In addition, NMS003 from Cambridge was genotyped based on inspection of its SNP profile, despite it not fulfilling the genomic coverage criteria for inclusion in our phylogenetic analysis (Supplementary Table 3), as its archaeological context makes it distinct from other Y. pestis-positive individuals from the same site (see Supplementary Note 1). As a result, SNP inspection classified it as potentially identical to other BD genomes (Supplementary Data 3). By contrast, certain isolates associated with the BD period are seemingly distinct. For example, TRP002 from Toulouse, which dates to 1347–1350 based on archaeological evidence, forms its own unique branch (Fig. 2; Supplementary Data 1). Qualitative assessment of eight unique SNPs in TRP002 with SNPEvaluation28 classified them as potential false-positives (see Methods, Supplementary Data 2). In addition, after visual inspection, all such variants appear in regions of the genome where reads from diverse sources seem to be mapping (Supplementary Fig. 17) and, therefore, were considered to be of exogenous origin. Similarly, we assessed one unique SNP identified in our re-analysis of the recently published OSL-1 genome from Oslo, Norway8 (Fig. 2). Visual inspection revealed it as a low-quality C-to-T transition that could be confined by aDNA damage (Supplementary Fig. 18). Finally, despite exclusion of BSS31 (Siena, Italy) from phylogenetic analysis, two previously identified unique SNPs in this genome were manually inspected, since they were presented as evidence for Y. pestis genetic diversity in Europe during the BD8. Importantly, BLASTn analysis of reads overlapping those regions (Supplementary Fig. 18, Supplementary Data 4 and 5) showed a 100% identity to environmental or other enteric bacterial species, but not to Y. pestis. We, hence, conclude that apart from LAI009 all reconstructed genomes associated with the initial pandemic wave have identical genotypes. In addition, we note that structural rearrangements could provide alternative means of genetic diversity. Although architectural differences are vastly abundant among modern Y. pestis genomes34, their assessment in ancient Y. pestis is limited by the short read aDNA data produced here. We find a number of genomes grouping with the previously described “post-BD” lineage together with published strains from Ellwangen (ELW098/549_O), Germany (1486–1630)9, and Marseille, France (1720–1722)14, which are descended from the European BD isolates (Fig. 2; Supplementary Data 1). Here, we identify the earliest evidence of this lineage in a 14th-century isolate from Manching-Pichl (MAN)13 (see Supplementary Note 1), which is followed by the more derived 15th- to 17th-century isolates from Starnberg (STA), Landsberg (LBG), Stans (STN) and Cambridge (NMS), as well as the 17th-century Brandenburg an der Havel (BRA)13 and London (BED), all of which provide further evidence for plague’s continuous presence in Europe after the BD. Of note, we retrieved eight nearly identical genomes from Stans (STN, maximum one SNP difference in two of eight genomes; mean SNP distance d = 0), and together with the four identical genomes from 17th-century London (BED) (d = 0), the five previously published nearly identical genomes from Marseille (OBS, maximum one SNP difference in one of five genomes, d = 0), and the seven identical BD isolates from various regions in Europe (d = 0), our results demonstrate low genetic diversity of the bacterium within local outbreaks and/or major epidemics of the second pandemic. In addition, we find that this “post-BD lineage” gave rise to (at least) two distinct clades within Europe, with the Ellwangen isolate being positioned closest to an apparent population split (Fig. 2). From this divergence, one clade gave rise to the strains associated with outbreaks in Germany and Switzerland (15th–17th century AD), and the second encompassed strains from 17th-century London (BED) and 18th-century Marseille (OBS). Notably, these two clades show dissimilar rates of substitution accumulation. For example, the mean SNP distance between the Ellwangen genome (ELW098/549_O) and the London (BED) genomes (d = 45) is double that observed between Ellwangen and Brandenburg (BRA, d = 22), despite an assumption of them being contemporaneous (early 17th century AD) based on archaeological dating (Fig. 2; Supplementary Table 1; Supplementary Note 1). Analysis of substitution rate variation in Y. pestis We used the Bayesian framework BEAST v1.8 in order to make an assessment of substitution rate variations across the genealogy of Branch 1 (n = 80), retaining high-quality second pandemic Y. pestis genomes and using available calibration points in our modern and ancient datasets (Supplementary Data 6). Previous studies have demonstrated that overdispersion among Y. pestis branch lengths is unlikely a result of natural selection, and have rather suggested a link between rate acceleration and geographic expansion of certain lineages during epidemic spread16,23. Our analysis based on the coalescent skyline model (Fig. 3, Supplementary Fig. 19) suggests an over 40-fold difference between the fastest and slowest substitution rates identified on Branch 1 (Fig. 3). In particular, we observe the fastest rates in three internal branches (Fig. 3). The first spans the genetic distance between the strains from Ellwangen (549_O) and London (BED), and supports the conflicting branch lengths of BED and BRA strains described earlier (Fig. 3 and Supplementary Data 7). The second is the branch leading to the 1.ANT strains isolated from Africa (Congo and Uganda) (Fig. 3 and Supplementary Data 8). The broad history of 1.ANT and the time period associated with its establishment in Africa are unknown, though an introduction from Eurasia has been hypothesised9,35. The third, which displays the fastest rate within the entire Branch 1, is the branch leading to 1.ORI isolates (Fig. 3 and Supplementary Data 9), which is associated with the global spread of Y. pestis via maritime routes during the third plague pandemic (1894–1950s)15,16. Our results, therefore, support the idea of faster substitution rates during epidemic spread, here particularly noticeable for lineages known to have expanded over wide geographic areas. 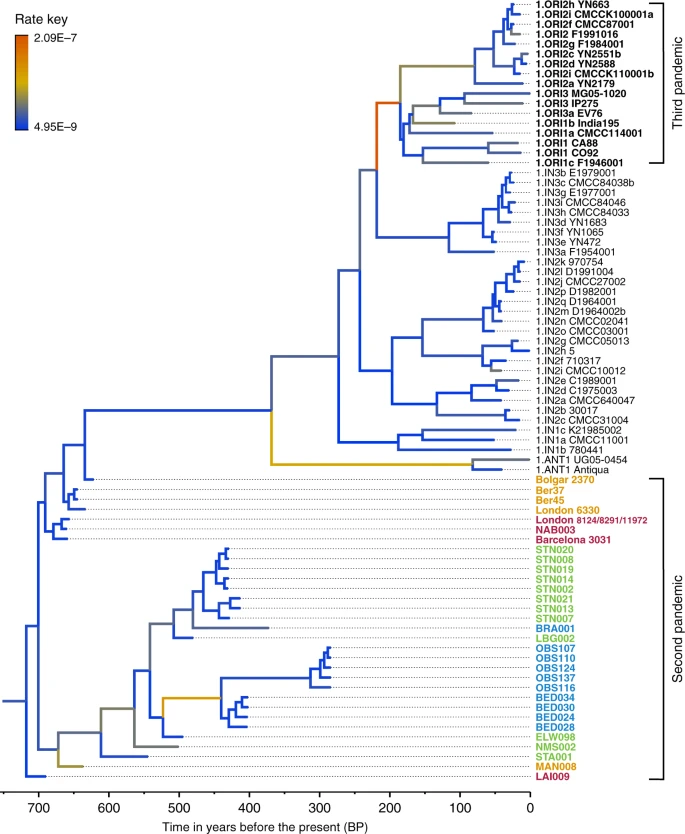 Fig. 3 Substitution rate variation across the Y. pestis Branch 1. The figure presents a maximum clade credibility (MCC) phylogenetic tree generated using BEAST v1.885 (rooted with 2.MED KIM10—outgroup not shown). The tree was viewed in FigTree v1.4 (http://tree.bio.ed.ac.uk/software/figtree/), and modified so that branch colours represent mean substitution rates (substitutions per site per year). The tree depicts the substitution rate variation across Branch 1 of the Y. pestis phylogeny, which ranges from 2.09E–7 (highest-red) to 4.95E–9 (lowest-blue) substitutions per site per year (see rate key). The isolates used for this analysis overlap with the ones used for the SNP and maximum likelihood phylogenetic analysis (see Supplementary Fig. 12), with the exception of the TRP002 and OSL1 genomes since their private SNP calls are likely affected by environmental contamination and other representative genomes exist in our dataset from the BD time period (1346–1353 AD). Labels of genomes associated with the second and third plague pandemics appear in bold. The mean substitution rate across the tree (including 2.MED KIM10) was calculated to 2.85E–8 substitutions per site per year. Lengths of branches are scaled to represent sample ages, and the depicted Branch 1 sequences are estimated to represent 731 years (95% HPD: 672–823) of Y. pestis evolution. The time scale is shown in years before the present (BP), where present denotes the most recently isolated modern Y. pestis strain (year 2005) |
|
|
|
Post by Admin on Oct 5, 2019 17:36:23 GMT
Analysis of virulence-associated genomic profiles To investigate the genomic profiles of all newly reconstructed genomes, we analysed the presence or absence of potential virulence-associated and evolutionary determinant genes located on the Y. pestis chromosome (Fig. 4a) and plasmids (Supplementary Fig. 20)36,37, in comparison to published representatives of ancient and modern strains. We find that the genetic profiles of some of the previously characterised historical strains are influenced by the capture design used for their retrieval. Specifically, the second pandemic genomes “Bolgar 2370” and “Barcelona 3031” (ref. 9) and the first pandemic genome “Altenerding 2148” (ref. 29) seem to lack coverage in certain Y. pestis-specific regions, since Yersinia pseudotuberculosis was previously used as a probe-design template for their enrichment9,29 (Fig. 4a). Regarding the newly reconstructed strains, we find that most possess all analysed genes with the exception of the New Churchyard (BED) and Marseille (OBS) strains that lack the magnesium transporter genes mgtB and mgtC, as well as the Cambridge (NMS002) strain that is lacking the inv gene (Fig. 4). While invasin is associated with epithelial colonisation of Y. pseudotuberculosis and Yersinia enterocolitica, it is known to have been inactivated in Y. pestis38. By contrast, magnesium transporters are considered vital for Y. pestis intracellular survival under low Mg2+ conditions39, such as those encountered within macrophage phagosomes. Specifically for Y. pestis, mgtB disruption has been associated with a decreased ability for macrophage invasion resulting in its attenuated virulence in mice40. Both mgtB and mgtC are present in all 233 modern Y. pestis genomes used in our comparative dataset. We explored these gene deletions in greater detail using BWA-MEM and identified them as part of a 49-kb missing region within the BED and OBS genomes (1,879,467–1,928,869 on CO92) (Fig. 4b, Supplementary Fig. 21) flanked by an IS100 element immediately following its downstream end, which is consistent with previously characterised disruptions or losses of Y. pestis genomic regions via insertion elements41. Apart from mgtB and mgtC, this region encompasses a set of 34 additional genes that code for both characterised and hypothetical proteins, most of which seem to be associated with phenotypic characteristics that appear inactivated in Y. pestis such as motility and chemotaxis as well as few genes associated with metabolism, structure synthesis and environmental stress response (Supplementary Fig. 21, Supplementary Table 4). In addition, the clade encompassing this deletion is associated with some of the late outbreaks of the second plague pandemic, i.e. during the 17th century in London, England (BED) (see Supplementary Note 1), and during the 18th-century Plague of Marseille, in France (OBS 1720–1722 AD)14, which was one of the last major epidemics that occurred in continental Europe42. Intriguingly, a nearly identical genomic deletion (45 kb), also including the mgtB and mgtC virulence-associated genes, was recently identified in ancient isolates from France (LVC, LSD)28 sequenced from victims of the first plague pandemic (6th–8th centuries AD)28. These genomes are described elsewhere and date within a wide temporal interval (550–650 AD), though based on existing data they appear to be the youngest first pandemic isolates sequenced to date28. 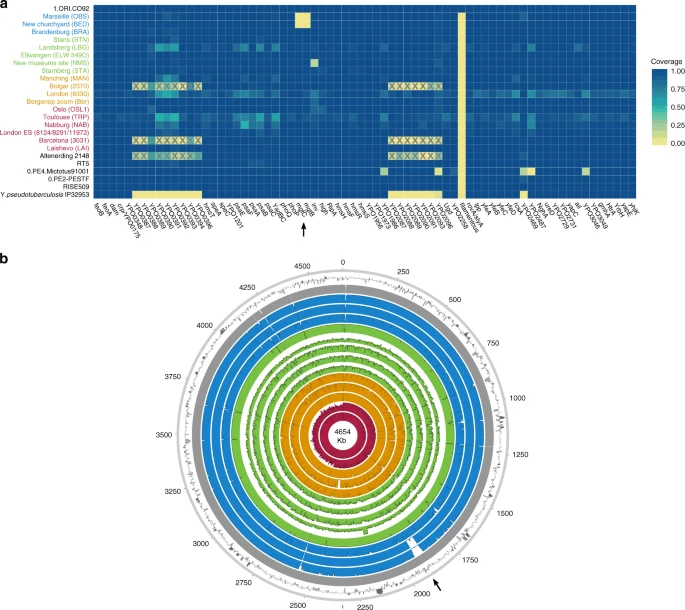 Fig. 4 Discussion A series of studies have sufficiently demonstrated the preservation of Y. pestis in ancient human remains from a wide temporal transect8,9,10,14,22,29,31,32,43. This study presents an extensive sampling of multiple European epidemic burials from the period between the 14th and 17th centuries in order to gain a more complete picture of Y. pestis’ genetic history during the second plague pandemic. Here, we nearly triple the amount of genomic data available from that time period (Fig. 1, Table 1 and Supplementary Table 3) and integration with existing datasets reveals key aspects regarding the initiation and progression of the second plague pandemic in Europe. Based on historical sources alone, it has been difficult to determine the time at which Y. pestis first reached different parts of western Russia7. A commonly accepted view dates its arrival in the southwest, particularly in cities of Astrakhan and Sarai, in 13461,44 with subsequent spread into southern Europe from the Crimean peninsula. On the other hand, the dispersal of plague into northwestern Russia (i.e. in the cities of Pskov and Novgorod7,44) may have followed an alternative route via the Baltic Sea, occurring at the end of the BD between 1351 and 13531,7,44. Such a notion of plague’s expansion from northern Europe eastwards is also supported by published ancient genomic data from the late 14th-century Middle Volga region of Russia9, though other scenarios may come to light with incorporation of additional genomic and historical data. Importantly, through analysis of our new strain from Laishevo (LAI009), which is phylogenetically ancestral to all second pandemic strains sequenced to date (Fig. 2), we provide evidence for the bacterium’s presence in the same region, ~2000 km northeast of the Crimean peninsula, prior to reaching southern Europe in 1347–13481 (currently represented by strains from Siena, Saint-Laurent-de-la-Cabrerisse, Barcelona and Toulouse8,9). These results suggest that the N07-derived SNP previously termed “p1”9 (Fig. 2, Supplementary Fig. 12), that is common to all other second pandemic strains, was likely acquired within Europe during the onset of the BD. In addition, given the proximity of the LAI009 genome to the N07 node often associated with the initiation of the BD (Fig. 2, Supplementary Fig. 12)23, further data will be necessary to accurately re-evaluate the geographic origin of Branch 1. Previous analyses have proposed East Asia as the mostly likely candidate for the N07 polytomy10,23 (Fig. 2). Such claims, however, cannot yet be verified given; (1) the apparent East Asian sampling bias of modern isolates23,45, (2) the lack of molecular evidence from East Asia dating to the early 14th century and (3) the scarcity of historical documentary sources from this region describing precise disease symptoms46. In addition, recently published modern Y. pestis genomes from Central Asia show a rich diversity in the local plague foci26,27, and further sampling from these regions has the potential to inform hypotheses on plague movement and evolution.
The identification of low genomic diversity during the initial wave of the second pandemic becomes particularly informative when attempting to reconstruct the spread of plague after 1353. Previous research based on climatic proxies12 as well as PCR47 and genomic8 data have proposed multiple introductory waves of Y. pestis into Europe as the main source for the post-BD outbreaks recorded until the 18th century. Here, using previously published8,9,10,14 and new whole-genome data from 20 archaeological sites, we identify that all genomes associated with post-BD outbreaks in Europe derived from a single ancestral strain that was present in southern, central, western and northern Europe during the BD. We, therefore, interpret the current data as supporting a single entry of Y. pestis during the BD, though additional interpretations may arise through the discovery of unsampled diversity in western Eurasia. Subsequent to its entry, we observe the formation of two sister lineages (Fig. 2). The first lineage is responsible for the bacterium’s possible eastward expansion after the BD. It contains strains from late-14th-century Bergen op Zoom, London (6330)10 and the city of Bolgar (2370)9, as well as extant strains from Africa (1.ANT)48, and, most importantly, a worldwide set of isolates associated with the third pandemic (1.ORI, 19th–20th centuries)15,16,23 (Fig. 2). The second, here termed the “post-BD lineage”, is characterised by a profound genomic diversity identified within Europe that seems to have been restricted to the second pandemic, as no modern descendants have been identified for this lineage to date. It is represented by historical genomes isolated from 14th- to 18th-century Germany (MAN, STA, ELW, LBG and BRA), Switzerland (STN), England (NMS, BED) and France (OBS) (Fig. 2), suggesting that it persisted in Europe or its vicinity and caused infections over a wide geographic range. The fact that this lineage has no identified modern descendants is likely related to the disappearance of plague from Europe in the 18th century, possibly due to extinction of local reservoirs, as previously suggested9. We find that the “post-BD lineage” gave rise to (at least) two distinct clades that separate the strains identified in Central Europe during the 15th–17th centuries, and those identified in 17th- to 18th-century England and France. Their distinction is corroborated not only by their genetic and geographic separation (Fig. 2), but also by potential differences in their genomic profiles (Fig. 4) and substitution rates (Fig. 3). The clade that exhibits a slower substitution rate is mainly represented by temporally and genetically closely related isolates from Germany and Switzerland (Fig. 2), which could indicate endemic circulation of the bacterium in that region. Such an observation may be compatible with the hypothesis of an Alpine rodent reservoir facilitating the spread of plague in Central Europe after the BD49, although a possible sampling bias should be noted since the majority of our data derive from this region. On the other hand, the clade that exhibits a faster substitution rate (Fig. 3) appears to have had a wider geographic distribution. Given that both Marseille and London were among the main maritime trade centres in Europe during that time, it is plausible that introduction of the disease in these areas occurred via ships50, although sources favouring local epidemic eruptions also exist51. Previous studies have demonstrated that transmission of Y. pestis via steamships during the 19th century played a significant role in initial introduction of the bacterium to several regions worldwide, such as in Madagascar where it persists until today15,16,52,53. As such, the possibility of maritime introductions of plague into London and Marseille during the second pandemic vastly expands the breadth of potential geographic source(s) for these strains. Nevertheless, the phylogenetic positioning of the BED and OBS genomes within the “post-BD lineage” and in relation to other second pandemic isolates suggests they arose within Europe or its vicinity. We identified a 49-kb deletion within both BED and OBS genomes (Fig. 4b), which caused the loss of two virulence-associated genes, mgtB and mgtC (Fig. 4a). This deletion could not be identified in other second pandemic or modern strains in our dataset (Supplementary Fig. 21). The inferred virulence potential of mgtB and mgtC genes is associated with intracellular survival of Y. pestis within macrophages40,54. Their co-expression has been shown to affect the virulence exerted by other pathogenic enterobacteria under laboratory conditions55,56 and both genes have been proposed as potential drug targets40,57. Moreover, the function of mgtB was shown to be temperature-dependent, being active at 37 °C but not at 20 °C58, suggesting its loss affects the bacterium in warm-blooded hosts. Intriguingly, a 45-kb deletion in the same region was identified in genomes associated with late outbreaks of the first plague pandemic (6th–8th century AD)28, which sets it as a candidate for convergent evolution and raises questions regarding its functional importance. Given that all genomes displaying this deletion were obtained from plague victims, including the Great Plague of Marseille (1720–1722 AD) that is known to have caused high mortality, its occurrence may not have reduced the pathogen’s virulence, particularly since genome decay is a well-established characteristic of Y. pestis evolution59,60. Nevertheless, since both lineages that show this deletion are likely extinct, its functional characterisation will be of importance to evaluate potential effects on maintenance in mammalian and arthropod hosts, in Europe, during the first and second pandemics. The second plague pandemic has arguably caused the highest levels of mortality of the three recorded plague pandemics1,61. It serves as a classic historical example of rapid infectious disease emergence, long-term local persistence and eventual extinction for reasons that are currently not understood. We have shown that extensive sampling of ancient Y. pestis genomic data can provide direct molecular evidence on the genetic relationships of strains present in Europe during that time. In addition, we provide relevant information regarding the initiation and progression of the second pandemic and suggest that a single source reservoir may be insufficient to explain the breadth of epidemics and Y. pestis’ genetic diversity in Europe during the 400-year course of the pandemic. Although certain key regions in western Eurasia remain under-sampled for ancient Y. pestis DNA, namely the eastern Mediterranean, Scandinavia and the Baltics, vast amounts of high-quality genomic data are becoming increasingly available. Their integration into disease modelling efforts, which consider vector transmission dynamics62,63, climatic12,64,65 and epidemiological data66, as well as a critical re-evaluation of historical records67, will become increasingly important for better understanding the second plague pandemic. Nature Communications volume 10, Article number: 4470 (2019) |
|
|
|
Post by Admin on Oct 6, 2019 18:28:17 GMT
Populations. The population of Romania is comprised mainly of Indo-European populations, among which Romanian speakers represent 88% of the population, whereas 3.2% of inhabitants are of Rroma ethnic background (www.recensamantromania.ro). After ethical approval by the Ethics Committee of the University of Craiova, Romania, informed consent was obtained for all volunteers and DNA samples were collected from individuals of European/Romanian or Rroma ethnic background. A population of individuals of Northwestern Indian descent, representing the geographic origin of the Rroma group (Fig. 1A), was also recruited.  Fig. 1. Geographic origin of the three populations studied. (A) European/Romanians and Rroma/Gipsy share the same location, even if the origin of the latter is in North India. (B) Plot of the populations under analysis according to the coordinates to the two main eigenvectors of smartpca (Eigensoft) analysis, in which each dot represents an individual. Individuals within the circles and the same color have been considered for the study; those of different colors represent false population allocation and those intermediate represent admixed individuals. ROM, nongypsy Romanians; INDI, individuals from North India; GYP, Rroma/Gypsies living in Romania. We assayed 196,524 single-nucleotide polymorphisms (SNPs) using the Illumina immunochip array (8) in all three populations. Analysis of genetic distance and principal component analysis between these populations based on nongenic, and thus presumably neutral, SNPs show clear differences between the three populations studied. Admixed individuals and erroneous self-assigned ancestry was examined using principal components analysis (PCA) implemented in eigensoft program (9) and plotted using multidimensional scaling (Fig. 1B). Individuals showing admixture ancestry or false allocation were excluded from further analysis. A plot of the first versus the second eigenvectors (Fig. 1B) shows a clear differentiation of the Rroma cluster of individuals from the Romanian and the Indian populations. However, Rroma are very close to Indians across eigenvector 1, in agreement with their evolutionary history. This indicates these population labels have a genetic basis and are not merely social constructs. Evolutionary Analysis Identifies Innate Immune Pathways and TLR1/TLR6/TLR10 Among Genes Under Common Selection Pressure in Europeans/Romanians and Rroma. To identify signals of positive selection shared between Europeans and Rroma but not present in the Indian population, we looked for shared signals of important genetic differentiation between these two populations with the Indian population, accompanied by the absence of genetic differentiation between them. Two tests were used: (i) Cross-Population Composite Likelihood Ratio (XP-CLR) (10), which is a test that aims to identify selective sweeps in a population by detecting important genetic differentiation in an extended genomic region by including information about linkage disequilibrium without requiring haplotype information, and (ii) TreeSelect test (11), which is a tree-based method that incorporates allele frequency information from all populations analyzed to increase power to detect selection and distinguishes which population has been under positive selection. A window was considered to show an extreme score if its summary statistic (maximum in the case of XP-CLR, mean in case of TreeSelect statistic) belonged to the 1% upper tail of the genome-wide summary statistic distribution. Therefore, for XP-CLR, we were interested in windows with the extreme 1% signal of population differentiation both between Rroma and Indians and between Europeans and Indians, as long as these windows did not belong to the 5% extreme distribution for the Rroma versus European comparison. For TreeSelect, we listed the windows belonging to the 1% upper tail of the distribution for Rromas and Romanians as long as they do not belong to the 5% upper tail of the distribution in Indians. Table 1 lists the genes contained in windows that fulfill these criteria, along with other genes highly significant in any of the tests in any of the three populations analyzed. Manhattan plots for XP-CLR and TreeSelect statistics are shown in Fig. 2 A and B, respectively, where the strong concordance between both tests can be seen.  Fig. 2. Manhattan plot of results of selection tests in Rroma, Romanians, and Indians using TreeSelect statistic (A) and XP-CLR statistic (B). Chromosomes ordered from chromosome 1 to chromosome 22. We investigated the overrepresentation of categories of genes detected to show similar selection signals in Rroma and Romanians and not in Indians, using Protein Analysis Through Evolutionary Relationships (PANTHER) (12) analysis. Table 2 shows the overrepresented molecular functions and biological processes with the contributing genes. The Toll-like receptor (TLR)/cytokine–mediated signaling pathway group, which comprises the genes TLR1, TLR6, and TLR10 (in the second cluster of Table 1), appears at the top of groups overrepresented with a P value = 0.00381. The finding of the TLR2 gene cluster as under positive selection is of great relevance in looking for convergent selection in Rromas and Romanians. To overcome a possible lack of power of detecting selection in Indians for this cluster, we sought derived allele frequency (DAF) of SNPs that shows signals of positive selection in this study. SNP rs4833103 has a DAF in Rroma of 0.3, in Romanians 0.5, and in Indians 0.02. For SNP imm_4_38475934, the DAF in Rroma is 0.05, in Romanians 0.04, and in Indians 0.007. This result suggests that the signals of positive selection can be attributed only to Rroma and Romanians. Moreover, population differentiation estimated by FST statistic shows that most of the SNPs within this cluster have high differentiation between Rroma and Indians and between Romanians and Indians but not between Rroma and Romanian. The case of SNP rs4833103 is of special interest; this SNP shows an FST between Rroma and Indians of 0.49, between Romanians and Indians 0.69, and between Rroma and Romanians 0.04 (Fig. S1), values of undoutable value for the present framework. Notably, this SNP (intergenic between TLR1 and TLR6) was reported to be associated with an expression quantitative trait loci of the expression of three genes, TLR1, TLR6, and TLR10, in lymphoblastoid cell lines (LCLs) (13). We also performed an additional analysis using genotype data from the Illumina Omni 2.5M Chip for the 1000 Genome Project for individuals (14) in an Indian (Gujarati) and European (Northern Europeans from Utah, CEU) population. XP-CLR statistic was used to detect selection in this Indian population. Results show that there is clear signal of selection in the European population (CEU) compared with the Indian (Gujarati) population, but no signal of selection was detected in this Indian population compared with the European population (Fig. S2 A and B). Interestingly, the other gene cluster detected (first row in Table 1), with four genes in chromosome 5, contains the well-known gene SLC45A2, described as being under positive selection in relation to skin pigmentation in Europeans (14). Other strong signals are for the BTNL2 gene locus in chromosome 6 coming from the TreeSelect test in Rroma and Romanian populations. This gene is highly polymorphic, with homology to the butyrophilin gene family, and is located at the border of the major histocompatibility complex (MHC) class I and class II regions in humans. This signal of positive selection may be due to the role of MHC in adaptation to pathogens in human history. Many other strong signals are shown in Fig. 2 A and B, however these signals are specific to one single population or show differentiation between Rroma and Romanians and cannot be caused by a convergent adaptation of the same evolutionary process in these two populations. |
|

