|
|
Post by Admin on Apr 13, 2020 22:16:27 GMT
Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China Introduction Coronaviruses are enveloped non-segmented positive-sense RNA viruses belonging to the family Coronaviridae and the order Nidovirales and broadly distributed in humans and other mammals.1 Although most human coronavirus infections are mild, the epidemics of the two betacoronaviruses, severe acute respiratory syndrome coronavirus (SARS-CoV)2, 3, 4 and Middle East respiratory syndrome coronavirus (MERS-CoV),5, 6 have caused more than 10 000 cumulative cases in the past two decades, with mortality rates of 10% for SARS-CoV and 37% for MERS-CoV.7, 8 The coronaviruses already identified might only be the tip of the iceberg, with potentially more novel and severe zoonotic events to be revealed. In December, 2019, a series of pneumonia cases of unknown cause emerged in Wuhan, Hubei, China, with clinical presentations greatly resembling viral pneumonia.9 Deep sequencing analysis from lower respiratory tract samples indicated a novel coronavirus, which was named 2019 novel coronavirus (2019-nCoV). Thus far, more than 800 confirmed cases, including in health-care workers, have been identified in Wuhan, and several exported cases have been confirmed in other provinces in China, and in Thailand, Japan, South Korea, and the USA. We aim to describe epidemiological, clinical, laboratory, and radiological characteristics, treatment, and outcomes of patients confirmed to have 2019-nCoV infection, and to compare the clinical features between intensive care unit (ICU) and non-ICU patients. We hope our study findings will inform the global community of the emergence of this novel coronavirus and its clinical features. Results By Jan 2, 2020, 41 admitted hospital patients were identified as laboratory-confirmed 2019-nCoV infection in Wuhan. 20 [49%]) of the 2019-nCoV-infected patients were aged 25–49 years, and 14 (34%) were aged 50–64 years (figure 1A). The median age of the patients was 49·0 years (IQR 41·0–58·0; table 1). In our cohort of the first 41 patients as of Jan 2, no children or adolescents were infected. Of the 41 patients, 13 (32%) were admitted to the ICU because they required high-flow nasal cannula or higher-level oxygen support measures to correct hypoxaemia. Most of the infected patients were men (30 [73%]); less than half had underlying diseases (13 [32%]), including diabetes (eight [20%]), hypertension (six [15%]), and cardiovascular disease (six [15%]).  Figure 1 Date of illness onset and age distribution of patients with laboratory-confirmed 2019-nCoV infection 27 (66%) patients had direct exposure to Huanan seafood market (figure 1B). Market exposure was similar between the patients with ICU care (nine [69%]) and those with non-ICU care (18 [64%]). The symptom onset date of the first patient identified was Dec 1, 2019. None of his family members developed fever or any respiratory symptoms. No epidemiological link was found between the first patient and later cases. The first fatal case, who had continuous exposure to the market, was admitted to hospital because of a 7-day history of fever, cough, and dyspnoea. 5 days after illness onset, his wife, a 53-year-old woman who had no known history of exposure to the market, also presented with pneumonia and was hospitalised in the isolation ward. The most common symptoms at onset of illness were fever (40 [98%] of 41 patients), cough (31 [76%]), and myalgia or fatigue (18 [44%]); less common symptoms were sputum production (11 [28%] of 39), headache (three [8%] of 38), haemoptysis (two [5%] of 39), and diarrhoea (one [3%] of 38; table 1). More than half of patients (22 [55%] of 40) developed dyspnoea. The median duration from illness onset to dyspnoea was 8·0 days (IQR 5·0–13·0). The median time from onset of symptoms to first hospital admission was 7·0 days (4·0–8·0), to shortness of breath was 8·0 days (5·0–13·0), to ARDS was 9·0 days (8·0–14·0), to mechanical ventilation was 10·5 days (7·0–14·0), and to ICU admission was 10·5 days (8·0–17·0; figure 2).  Figure 2 Timeline of 2019-nCoV cases after onset of illness The blood counts of patients on admission showed leucopenia (white blood cell count less than 4 × 109/L; ten [25%] of 40 patients) and lymphopenia (lymphocyte count <1·0 × 109/L; 26 [63%] patients; table 2). Prothrombin time and D-dimer level on admission were higher in ICU patients (median prothrombin time 12·2 s [IQR 11·2–13·4]; median D-dimer level 2·4 mg/L [0·6–14·4]) than non-ICU patients (median prothrombin time 10·7 s [9·8–12·1], p=0·012; median D-dimer level 0·5 mg/L [0·3–0·8], p=0·0042). Levels of aspartate aminotransferase were increased in 15 (37%) of 41 patients, including eight (62%) of 13 ICU patients and seven (25%) of 28 non-ICU patients. Hypersensitive troponin I (hs-cTnI) was increased substantially in five patients, in whom the diagnosis of virus-related cardiac injury was made. |
|
|
|
Post by Admin on Apr 14, 2020 2:47:05 GMT
Most patients had normal serum levels of procalcitonin on admission (procalcitonin <0·1 ng/mL; 27 [69%] patients; table 2). Four ICU patients developed secondary infections. Three of the four patients with secondary infection had procalcitonin greater than 0·5 ng/mL (0·69 ng/mL, 1·46 ng/mL, and 6·48 ng/mL). On admission, abnormalities in chest CT images were detected among all patients. Of the 41 patients, 40 (98%) had bilateral involvement (table 2). The typical findings of chest CT images of ICU patients on admission were bilateral multiple lobular and subsegmental areas of consolidation (figure 3A). The representative chest CT findings of non-ICU patients showed bilateral ground-glass opacity and subsegmental areas of consolidation (figure 3B). Later chest CT images showed bilateral ground-glass opacity, whereas the consolidation had been resolved (figure 3C).  Figure 3 Chest CT images Initial plasma IL1B, IL1RA, IL7, IL8, IL9, IL10, basic FGF, GCSF, GMCSF, IFNγ, IP10, MCP1, MIP1A, MIP1B, PDGF, TNFα, and VEGF concentrations were higher in both ICU patients and non-ICU patients than in healthy adults (appendix pp 6–7). Plasma levels of IL5, IL12p70, IL15, Eotaxin, and RANTES were similar between healthy adults and patients infected with 2019-nCoV. Further comparison between ICU and non-ICU patients showed that plasma concentrations of IL2, IL7, IL10, GCSF, IP10, MCP1, MIP1A, and TNFα were higher in ICU patients than non-ICU patients. All patients had pneumonia. Common complications included ARDS (12 [29%] of 41 patients), followed by RNAaemia (six [15%] patients), acute cardiac injury (five [12%] patients), and secondary infection (four [10%] patients; table 3). Invasive mechanical ventilation was required in four (10%) patients, with two of them (5%) had refractory hypoxaemia and received extracorporeal membrane oxygenation as salvage therapy. All patients were administered with empirical antibiotic treatment, and 38 (93%) patients received antiviral therapy (oseltamivir). Additionally, nine (22%) patients were given systematic corticosteroids. A comparison of clinical features between patients who received and did not receive systematic corticosteroids is in the appendix (pp 1–5). As of Jan 22, 2020, 28 (68%) of 41 patients have been discharged and six (15%) patients have died. Fitness for discharge was based on abatement of fever for at least 10 days, with improvement of chest radiographic evidence and viral clearance in respiratory samples from upper respiratory tract. Discussion We report here a cohort of 41 patients with laboratory-confirmed 2019-nCoV infection. Patients had serious, sometimes fatal, pneumonia and were admitted to the designated hospital in Wuhan, China, by Jan 2, 2020. Clinical presentations greatly resemble SARS-CoV. Patients with severe illness developed ARDS and required ICU admission and oxygen therapy. The time between hospital admission and ARDS was as short as 2 days. At this stage, the mortality rate is high for 2019-nCoV, because six (15%) of 41 patients in this cohort died. The number of deaths is rising quickly. As of Jan 24, 2020, 835 laboratory-confirmed 2019-nCoV infections were reported in China, with 25 fatal cases. Reports have been released of exported cases in many provinces in China, and in other countries; some health-care workers have also been infected in Wuhan. Taken together, evidence so far indicates human transmission for 2019-nCoV. We are concerned that 2019-nCoV could have acquired the ability for efficient human transmission.19 Airborne precautions, such as a fit-tested N95 respirator, and other personal protective equipment are strongly recommended. To prevent further spread of the disease in health-care settings that are caring for patients infected with 2019-nCoV, onset of fever and respiratory symptoms should be closely monitored among health-care workers. Testing of respiratory specimens should be done immediately once a diagnosis is suspected. Serum antibodies should be tested among health-care workers before and after their exposure to 2019-nCoV for identification of asymptomatic infections. Similarities of clinical features between 2019-nCoV and previous betacoronavirus infections have been noted. In this cohort, most patients presented with fever, dry cough, dyspnoea, and bilateral ground-glass opacities on chest CT scans. These features of 2019-nCoV infection bear some resemblance to SARS-CoV and MERS-CoV infections.20, 21 However, few patients with 2019-nCoV infection had prominent upper respiratory tract signs and symptoms (eg, rhinorrhoea, sneezing, or sore throat), indicating that the target cells might be located in the lower airway. Furthermore, 2019-nCoV patients rarely developed intestinal signs and symptoms (eg, diarrhoea), whereas about 20–25% of patients with MERS-CoV or SARS-CoV infection had diarrhoea.21 Faecal and urine samples should be tested to exclude a potential alternative route of transmission that is unknown at this stage. The pathophysiology of unusually high pathogenicity for SARS-CoV or MERS-CoV has not been completely understood. Early studies have shown that increased amounts of proinflammatory cytokines in serum (eg, IL1B, IL6, IL12, IFNγ, IP10, and MCP1) were associated with pulmonary inflammation and extensive lung damage in SARS patients.22 MERS-CoV infection was also reported to induce increased concentrations of proinflammatory cytokines (IFNγ, TNFα, IL15, and IL17).23 We noted that patients infected with 2019-nCoV also had high amounts of IL1B, IFNγ, IP10, and MCP1, probably leading to activated T-helper-1 (Th1) cell responses. Moreover, patients requiring ICU admission had higher concentrations of GCSF, IP10, MCP1, MIP1A, and TNFα than did those not requiring ICU admission, suggesting that the cytokine storm was associated with disease severity. However, 2019-nCoV infection also initiated increased secretion of T-helper-2 (Th2) cytokines (eg, IL4 and IL10) that suppress inflammation, which differs from SARS-CoV infection.22 Further studies are necessary to characterise the Th1 and Th2 responses in 2019-nCoV infection and to elucidate the pathogenesis. Autopsy or biopsy studies would be the key to understand the disease. In view of the high amount of cytokines induced by SARS-CoV,22, 24 MERS-CoV,25, 26 and 2019-nCoV infections, corticosteroids were used frequently for treatment of patients with severe illness, for possible benefit by reducing inflammatory-induced lung injury. However, current evidence in patients with SARS and MERS suggests that receiving corticosteroids did not have an effect on mortality, but rather delayed viral clearance.27, 28, 29 Therefore, corticosteroids should not be routinely given systemically, according to WHO interim guidance.30 Among our cohort of 41 laboratory-confirmed patients with 2019-nCoV infection, corticosteroids were given to very few non-ICU cases, and low-to-moderate dose of corticosteroids were given to less than half of severely ill patients with ARDS. Further evidence is urgently needed to assess whether systematic corticosteroid treatment is beneficial or harmful for patients infected with 2019-nCoV. No antiviral treatment for coronavirus infection has been proven to be effective. In a historical control study,31 the combination of lopinavir and ritonavir among SARS-CoV patients was associated with substantial clinical benefit (fewer adverse clinical outcomes). Arabi and colleagues initiated a placebo-controlled trial of interferon beta-1b, lopinavir, and ritonavir among patients with MERS infection in Saudi Arabia.32 Preclinical evidence showed the potent efficacy of remdesivir (a broad-spectrum antiviral nucleotide prodrug) to treat MERS-CoV and SARS-CoV infections.33, 34 As 2019-nCoV is an emerging virus, an effective treatment has not been developed for disease resulting from this virus. Since the combination of lopinavir and ritonavir was already available in the designated hospital, a randomised controlled trial has been initiated quickly to assess the efficacy and safety of combined use of lopinavir and ritonavir in patients hospitalised with 2019-nCoV infection. Our study has some limitations. First, for most of the 41 patients, the diagnosis was confirmed with lower respiratory tract specimens and no paired nasopharyngeal swabs were obtained to investigate the difference in the viral RNA detection rate between upper and lower respiratory tract specimens. Serological detection was not done to look for 2019-nCoV antibody rises in 18 patients with undetectable viral RNA. Second, with the limited number of cases, it is difficult to assess host risk factors for disease severity and mortality with multivariable-adjusted methods. This is a modest-sized case series of patients admitted to hospital; collection of standardised data for a larger cohort would help to further define the clinical presentation, natural history, and risk factors. Further studies in outpatient, primary care, or community settings are needed to get a full picture of the spectrum of clinical severity. At the same time, finding of statistical tests and p values should be interpreted with caution, and non-significant p values do not necessarily rule out difference between ICU and non-ICU patients. Third, since the causative pathogen has just been identified, kinetics of viral load and antibody titres were not available. Finally, the potential exposure bias in our study might account for why no paediatric or adolescent patients were reported in this cohort. More effort should be made to answer these questions in future studies. Both SARS-CoV and MERS-CoV were believed to originate in bats, and these infections were transmitted directly to humans from market civets and dromedary camels, respectively.35 Extensive research on SARS-CoV and MERS-CoV has driven the discovery of many SARS-like and MERS-like coronaviruses in bats. In 2013, Ge and colleagues36 reported the whole genome sequence of a SARS-like coronavirus in bats with that ability to use human ACE2 as a receptor, thus having replication potentials in human cells.37 2019-nCoV still needs to be studied deeply in case it becomes a global health threat. Reliable quick pathogen tests and feasible differential diagnosis based on clinical description are crucial for clinicians in their first contact with suspected patients. Because of the pandemic potential of 2019-nCoV, careful surveillance is essential to monitor its future host adaption, viral evolution, infectivity, transmissibility, and pathogenicity. Published:January 24, 2020 DOI:https://doi.org/10.1016/S0140-6736(20)30183-5 |
|
|
|
Post by Admin on Apr 14, 2020 20:53:15 GMT
Phylogenetic network analysis of SARS-CoV-2 genomes Peter Forster, Lucy Forster, Colin Renfrew, and Michael Forster PNAS first published April 8, 2020 doi.org/10.1073/pnas.2004999117Abstract In a phylogenetic network analysis of 160 complete human severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2) genomes, we find three central variants distinguished by amino acid changes, which we have named A, B, and C, with A being the ancestral type according to the bat outgroup coronavirus. The A and C types are found in significant proportions outside East Asia, that is, in Europeans and Americans. In contrast, the B type is the most common type in East Asia, and its ancestral genome appears not to have spread outside East Asia without first mutating into derived B types, pointing to founder effects or immunological or environmental resistance against this type outside Asia. The network faithfully traces routes of infections for documented coronavirus disease 2019 (COVID-19) cases, indicating that phylogenetic networks can likewise be successfully used to help trace undocumented COVID-19 infection sources, which can then be quarantined to prevent recurrent spread of the disease worldwide. The search for human origins seemed to take a step forward with the publication of the global human mitochondrial DNA tree (1). It soon turned out, however, that the tree-building method did not facilitate an unambiguous interpretation of the data. This motivated the development, in the early 1990s, of phylogenetic network methods which are capable of enabling the visualization of a multitude of optimal trees (2, 3). This network approach, based on mitochondrial and Y chromosomal data, allowed us to reconstruct the prehistoric population movements which colonized the planet (4, 5). The phylogenetic network approach from 2003 onward then found application in the reconstruction of language prehistory (6). It is now timely to apply the phylogenetic network approach to virological data to explore how this method can contribute to an understanding of coronavirus evolution. In early March 2020, the GISAID database (https://www.gisaid.org/) contained a compilation of 253 severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) complete and partial genomes contributed by clinicians and researchers from across the world since December 2019. To understand the evolution of this virus within humans, and to assist in tracing infection pathways and designing preventive strategies, we here present a phylogenetic network of 160 largely complete SARS-Cov-2 genomes (Fig. 1).  Fig. 1. Phylogenetic network of 160 SARS-CoV-2 genomes. Node A is the root cluster obtained with the bat (R. affinis) coronavirus isolate BatCoVRaTG13 from Yunnan Province. Circle areas are proportional to the number of taxa, and each notch on the links represents a mutated nucleotide position. The sequence range under consideration is 56 to 29,797, with nucleotide position (np) numbering according to the Wuhan 1 reference sequence (8). The median-joining network algorithm (2) and the Steiner algorithm (9) were used, both implemented in the software package Network5011CS (https://www.fluxus-engineering.com/), with the parameter epsilon set to zero, generating this network containing 288 most-parsimonious trees of length 229 mutations. The reticulations are mainly caused by recurrent mutations at np11083. The 161 taxa (160 human viruses and one bat virus) yield 101 distinct genomic sequences. The phylogenetic diagram is available for detailed scrutiny in A0 poster format (SI Appendix, Fig. S5) and in the free Network download files. Zhou et al. (7) recently reported a closely related bat coronavirus, with 96.2% sequence similarity to the human virus. We use this bat virus as an outgroup, resulting in the root of the network being placed in a cluster of lineages which we have labeled “A.” Overall, the network, as expected in an ongoing outbreak, shows ancestral viral genomes existing alongside their newly mutated daughter genomes. There are two subclusters of A which are distinguished by the synonymous mutation T29095C. In the T-allele subcluster, four Chinese individuals (from the southern coastal Chinese province of Guangdong) carry the ancestral genome, while three Japanese and two American patients differ from it by a number of mutations. These American patients are reported to have had a history of residence in the presumed source of the outbreak in Wuhan. The C-allele subcluster sports relatively long mutational branches and includes five individuals from Wuhan, two of which are represented in the ancestral node, and eight other East Asians from China and adjacent countries. It is noteworthy that nearly half (15/33) of the types in this subcluster, however, are found outside East Asia, mainly in the United States and Australia. Two derived network nodes are striking in terms of the number of individuals included in the nodal type and in mutational branches radiating from these nodes. We have labeled these phylogenetic clusters B and C. For type B, all but 19 of the 93 type B genomes were sampled in Wuhan (n = 22), in other parts of eastern China (n = 31), and, sporadically, in adjacent Asian countries (n = 21). Outside of East Asia, 10 B-types were found in viral genomes from the United States and Canada, one in Mexico, four in France, two in Germany, and one each in Italy and Australia. Node B is derived from A by two mutations: the synonymous mutation T8782C and the nonsynonymous mutation C28144T changing a leucine to a serine. Cluster B is striking with regard to mutational branch lengths: While the ancestral B type is monopolized (26/26 genomes) by East Asians, every single (19/19) B-type genome outside of Asia has evolved mutations. This phenomenon does not appear to be due to the month-long time lag and concomitant mutation rate acting on the viral genome before it spread outside of China (Dataset S1, Supplementary Table 2). A complex founder scenario is one possibility, and a different explanation worth considering is that the ancestral Wuhan B-type virus is immunologically or environmentally adapted to a large section of the East Asian population, and may need to mutate to overcome resistance outside East Asia. Type C differs from its parent type B by the nonsynonymous mutation G26144T which changes a glycine to a valine. In the dataset, this is the major European type (n = 11), with representatives in France, Italy, Sweden, and England, and in California and Brazil. It is absent in the mainland Chinese sample, but evident in Singapore (n = 5) and also found in Hong Kong, Taiwan, and South Korea. One practical application of the phylogenetic network is to reconstruct infection paths where they are unknown and pose a public health risk. The following cases where the infection history is well documented may serve as illustrations (SI Appendix). On 25 February 2020, the first Brazilian was reported to have been infected following a visit to Italy, and the network algorithm reflects this with a mutational link between an Italian and his Brazilian viral genome in cluster C (SI Appendix, Fig. S1). In another case, a man from Ontario had traveled from Wuhan in central China to Guangdong in southern China and then returned to Canada, where he fell ill and was conclusively diagnosed with coronavirus disease 2019 (COVID-19) on 27 January 2020. In the phylogenetic network (SI Appendix, Fig. S2), his virus genome branches from a reconstructed ancestral node, with derived virus variants in Foshan and Shenzhen (both in Guangdong province), in agreement with his travel history. His virus genome now coexists with those of other infected North Americans (one Canadian and two Californians) who evidently share a common viral genealogy. The case of the single Mexican viral genome in the network is a documented infection diagnosed on 28 February 2020 in a Mexican traveler to Italy. Not only does the network confirm the Italian origin of the Mexican virus (SI Appendix, Fig. S3), but it also implies that this Italian virus derives from the first documented German infection on 27 January 2020 in an employee working for the Webasto company in Munich, who, in turn, had contracted the infection from a Chinese colleague in Shanghai who had received a visit by her parents from Wuhan. This viral journey from Wuhan to Mexico, lasting a month, is documented by 10 mutations in the phylogenetic network. This viral network is a snapshot of the early stages of an epidemic before the phylogeny becomes obscured by subsequent migration and mutation. The question may be asked whether the rooting of the viral evolution can be achieved at this early stage by using the oldest available sampled genome as a root. As SI Appendix, Fig. S4 shows, however, the first virus genome that was sampled on 24 December 2019 already is distant from the root type according to the bat coronavirus outgroup rooting. The described core mutations have been confirmed by a variety of contributing laboratories and sequencing platforms and can be considered reliable. The phylogeographic patterns in the network are potentially affected by distinctive migratory histories, founder events, and sample size. Nevertheless, it would be prudent to consider the possibility that mutational variants might modulate the clinical presentation and spread of the disease. The phylogenetic classification provided here may be used to rule out or confirm such effects when evaluating clinical and epidemiological outcomes of SARS-CoV-2 infection, and when designing treatment and, eventually, vaccines. |
|
|
|
Post by Admin on Apr 15, 2020 7:04:27 GMT
Scientists say they have discovered the first evidence of a “significant” mutation of the coronavirus — raising concerns that strides made toward a vaccine so far could become “futile,” according to a new study.  The researchers, who isolated a strain of the virus from a sample collected in India in January, said the mutation appeared to make the bug less able to bind to a receptor on human cells called ACE2, an enzyme found in the lungs. The discovery of this mutation “raises the alarm that the ongoing vaccine development may become futile in future epidemic if more mutations were identified,” the researchers said, according to Newsweek. The study, which was published on biorxiv.org on Saturday, has not yet been peer-reviewed. However, the team — led by the National Changhua University of Education in Taiwan in collaboration with Murdoch University in Australia — added that SARS-CoV-2, the virus which causes COVID-19, has a low mutation rate. “We confirmed that SARS-CoV-2 has a relatively low mutation rate but also proved that novel mutation with varied virulence and immune characteristics have already emerged,” they said, according to the outlet. But Jenna Macciochi, a lecturer in immunology at the University of Sussex who did not work on the study, told Newsweek that although the finding is important to monitor, she doesn’t believe vaccination efforts are hindered. “Small mutations would be expected with any virus. The emerged mutation in this report appears to reduce binding to ACE2 meaning less virulence which could potentially mean less ability to infect. But as this is an isolated report this doesn’t necessarily mean vaccine attempts are futile.” Analysis of the mutation dynamics of SARS-CoV-2 reveals the spread history and emergence of RBD mutant with lower ACE2 binding affinity YONG JIA, Gangxu Shen, Yujuan Zhang, Keng-Shiang Huang, Hsing-Ying Ho, Wei-Shio Hor, Chih-Hui Yang, Chengdao Li, Wei-Lung Wang Abstract Monitoring the mutation dynamics of SARS-CoV-2 is critical for the development of effective approaches to contain the pathogen. By analyzing 106 SARS-CoV-2 and 39 SARS genome sequences, we provided direct genetic evidence that SARS-CoV-2 has a much lower mutation rate than SARS. Minimum Evolution phylogeny analysis revealed the putative original status of SARS-CoV-2 and the early-stage spread history. The discrepant phylogenies for the spike protein and its receptor binding domain proved a previously reported structural rearrangement prior to the emergence of SARS-CoV-2. Despite that we found the spike glycoprotein of SARS-CoV-2 is particularly more conserved, we identified a mutation that leads to weaker receptor binding capability, which concerns a SARS-CoV-2 sample collected on 27th January 2020 from India. This represents the first report of a significant SARS-CoV-2 mutant, and raises the alarm that the ongoing vaccine development may become futile in future epidemic if more mutations were identified. doi: doi.org/10.1101/2020.04.09.034942 |
|
|
|
Post by Admin on May 28, 2020 0:39:44 GMT
The first round of results from an immunological study of 149 people who have recovered from COVID-19 show that although the amount of antibodies they generated varies widely, most individuals had generated at least some that were intrinsically capable of neutralizing the SARS-CoV-2 virus. Antibodies vary widely in their efficacy. While many may latch on to the virus, only some are truly "neutralizing," meaning that they actually block the virus from entering the cells. Since April 1, a team of immunologists, medical scientists, and virologists, has been collecting blood samples from volunteers who have recovered from COVID-19. The majority of the samples they have studied showed poor to modest "neutralizing activity," indicating a weak antibody response. However, a closer look revealed everyone's immune system is capable of generating effective antibodies—just not necessarily enough of them. Even when neutralizing antibodies were not present in an individual's serum in large quantities, researchers could find some rare immune cells that make them. "This suggests just about everybody can do this, which is very good news for vaccines," says Michel C. Nussenzweig, head of the Laboratory of Molecular Immunology at Rockefeller. "It means if you were able to create a vaccine that elicits these particular antibodies, then the vaccine is likely to be effective and work for a lot of people." Moreover, the researchers identified three distinct antibodies that were shown to be the most potent of the bunch in neutralizing the virus. They are working to develop them further into therapeutic and preventive drugs. The findings are shared on BioRxiv ahead of submission to peer-reviewed scientific journals. Nussenzweig's collaborators include, Davide F. Robbiani, Marina Caskey, Paul Bieniasz, Theodora Hatziioannou, and Charles M. Rice. 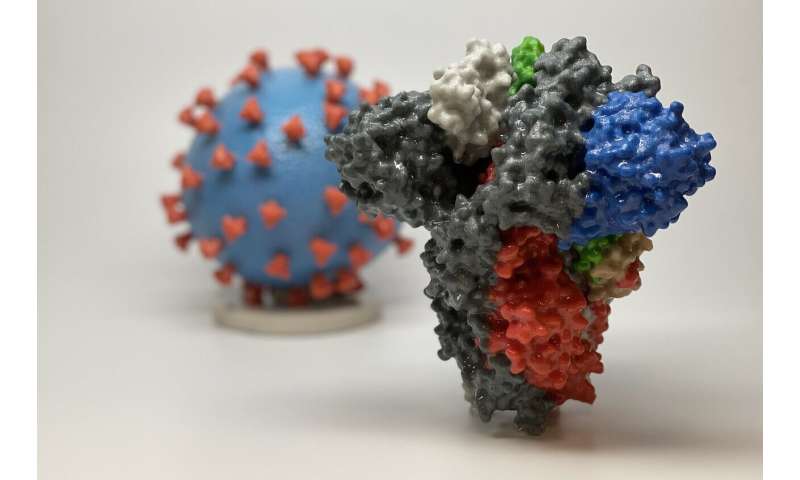 Antibody spectrum From the beginning of April and over five weeks, 149 people who had recovered from COVID-19 visited The Rockefeller Hospital to donate plasma, the portion of the blood that contains the antibodies, and the immune B-cells that produce them. The participants had experienced symptoms for an average of 12 days, and had their first symptoms on average 39 days before plasma donation. Bieniasz and Hatziioannou's team used an essay they had developed to test the neutralizing activity of the plasma samples. This involved mixing the plasma with a pseudo SARS-CoV-2 virus and measuring how well this mixture could still infect human cells in a dish. In 33 percent of donors, the neutralizing activity of plasma was below detectable levels. It's possible that for many in this group, their immune system's first line of defense had resolved the infection quickly, before the antibody-producing cells were called in. The majority of the plasma samples showed poor to modest neutralizing activity. And for 1 percent of donors it was remarkably high. "Like in other diseases, everyone responds differently," says Robbiani, research associate professor at Laboratory of Molecular Immunology. "Some people have poor response, some average. And then there is a fraction of people that are exceptional responders." Those "elite" responders are crucial to the team's plans. The high numbers of neutralizing antibodies in their serum makes it possible for researcher to catch the rare B cells that make them. They can then clone the antibodies from those cells, and use them to emulate the same strong defense in other people. Out of the numerous antibodies generated by elite responders who had the best performing plasma, the team identified 40 that neutralized the virus, and zeroed in on three that could do so even at very low concentrations. The team has cloned these most potent antibodies and is now working to develop them for clinical use. Neutralizing antibodies found in this study bind to at least three distinct sites on the receptor-binding domain (RBD) subunit of the spike protein, which is what SARS-CoV-2 uses to gain entry to host cells. A second look at the low-performing plasma samples revealed they also contained these RBD-binding antibodies, albeit in small quantities. |
|

