Post by Admin on May 9, 2020 20:16:20 GMT
Published: 03 February 2020
A pneumonia outbreak associated with a new coronavirus of probable bat origin
Peng Zhou, Xing-Lou Yang, […]Zheng-Li Shi
Nature volume 579, pages270–273(2020)
Abstract
Since the outbreak of severe acute respiratory syndrome (SARS) 18 years ago, a large number of SARS-related coronaviruses (SARSr-CoVs) have been discovered in their natural reservoir host, bats1,2,3,4. Previous studies have shown that some bat SARSr-CoVs have the potential to infect humans5,6,7. Here we report the identification and characterization of a new coronavirus (2019-nCoV), which caused an epidemic of acute respiratory syndrome in humans in Wuhan, China. The epidemic, which started on 12 December 2019, had caused 2,794 laboratory-confirmed infections including 80 deaths by 26 January 2020. Full-length genome sequences were obtained from five patients at an early stage of the outbreak. The sequences are almost identical and share 79.6% sequence identity to SARS-CoV. Furthermore, we show that 2019-nCoV is 96% identical at the whole-genome level to a bat coronavirus. Pairwise protein sequence analysis of seven conserved non-structural proteins domains show that this virus belongs to the species of SARSr-CoV. In addition, 2019-nCoV virus isolated from the bronchoalveolar lavage fluid of a critically ill patient could be neutralized by sera from several patients. Notably, we confirmed that 2019-nCoV uses the same cell entry receptor—angiotensin converting enzyme II (ACE2)—as SARS-CoV.
Download PDF
Main
Coronaviruses have caused two large-scale pandemics in the past two decades, SARS and Middle East respiratory syndrome (MERS)8,9. It has generally been thought that SARSr-CoV—which is mainly found in bats—could cause a future disease outbreak10,11. Here we report on a series of cases caused by an unidentified pneumonia disease outbreak in Wuhan, Hubei province, central China. This disease outbreak—which started from a local seafood market—has grown substantially to infect 2,761 people in China, is associated with 80 deaths and has led to the infection of 33 people in 10 additional countries as of 26 January 202012. Typical clinical symptoms of these patients are fever, dry cough, breathing difficulties (dyspnoea), headache and pneumonia. Disease onset may result in progressive respiratory failure owing to alveolar damage (as observed by transverse chest computerized-tomography images) and even death. The disease was determined to be caused by virus-induced pneumonia by clinicians according to clinical symptoms and other criteria, including a rise in body temperature, decreases in the number of lymphocytes and white blood cells (although levels of the latter were sometimes normal), new pulmonary infiltrates on chest radiography and no obvious improvement after treatment with antibiotics for three days. It appears that most of the early cases had contact history with the original seafood market; however, the disease has now progressed to be transmitted by human-to-human contact.
Samples from seven patients with severe pneumonia (six of whom are sellers or deliverymen from the seafood market), who were admitted to the intensive care unit of Wuhan Jin Yin-Tan Hospital at the beginning of the outbreak, were sent to the laboratory at the Wuhan Institute of Virology (WIV) for the diagnosis of the causative pathogen (Extended Data Table 1). As a laboratory investigating CoV, we first used pan-CoV PCR primers to test these samples13, given that the outbreak occurred in winter and in a market—the same environment as SARS infections. We found five samples to be PCR-positive for CoVs. One sample (WIV04), collected from the bronchoalveolar lavage fluid (BALF), was analysed by metagenomics analysis using next-generation sequencing to identify potential aetiological agents. Of the 10,038,758 total reads—of which 1,582 total reads were retained after filtering of reads from the human genome—1,378 (87.1%) sequences matched the sequence of SARSr-CoV (Fig. 1a). By de novo assembly and targeted PCR, we obtained a 29,891-base-pair CoV genome that shared 79.6% sequence identity to SARS-CoV BJ01 (GenBank accession number AY278488.2). High genome coverage was obtained by remapping the total reads to this genome (Extended Data Fig. 1). This sequence has been submitted to GISAID (https://www.gisaid.org/) (accession number EPI_ISL_402124). Following the name given by the World Health Organization (WHO), we tentatively call it novel coronavirus 2019 (2019-nCoV). Four more full-length genome sequences of 2019-nCoV (WIV02, WIV05, WIV06 and WIV07) (GISAID accession numbers EPI_ISL_402127–402130) that were more than 99.9% identical to each other were subsequently obtained from four additional patients using next-generation sequencing and PCR (Extended Data Table 2).
Fig. 1: Genome characterization of 2019-nCoV.
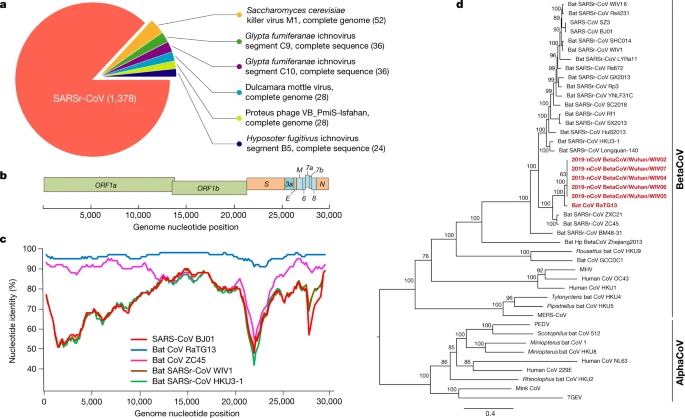
a, Metagenomics analysis of next-generation sequencing of BALF from patient ICU06. b, Genomic organization of 2019-nCoV WIV04. M, membrane. c, Similarity plot based on the full-length genome sequence of 2019-nCoV WIV04. Full-length genome sequences of SARS-CoV BJ01, bat SARSr-CoV WIV1, bat coronavirus RaTG13 and ZC45 were used as reference sequences. d, Phylogenetic tree based on nucleotide sequences of complete genomes of coronaviruses. MHV, murine hepatitis virus; PEDV, porcine epidemic diarrhoea virus; TGEV, porcine transmissible gastroenteritis virus.The scale bars represent 0.1 substitutions per nucleotide position. Descriptions of the settings and software that was used are included in the Methods.
The virus genome consists of six major open-reading frames (ORFs) that are common to coronaviruses and a number of other accessory genes (Fig. 1b). Further analysis indicates that some of the 2019-nCoV genes shared less than 80% nucleotide sequence identity to SARS-CoV. However, the amino acid sequences of the seven conserved replicase domains in ORF1ab that were used for CoV species classification were 94.4% identical between 2019-nCoV and SARS-CoV, suggesting that the two viruses belong to the same species, SARSr-CoV.
We then found that a short region of RNA-dependent RNA polymerase (RdRp) from a bat coronavirus (BatCoV RaTG13)—which was previously detected in Rhinolophus affinis from Yunnan province—showed high sequence identity to 2019-nCoV. We carried out full-length sequencing on this RNA sample (GISAID accession number EPI_ISL_402131). Simplot analysis showed that 2019-nCoV was highly similar throughout the genome to RaTG13 (Fig. 1c), with an overall genome sequence identity of 96.2%. Using the aligned genome sequences of 2019-nCoV, RaTG13, SARS-CoV and previously reported bat SARSr-CoVs, no evidence for recombination events was detected in the genome of 2019-nCoV. Phylogenetic analysis of the full-length genome and the gene sequences of RdRp and spike (S) showed that—for all sequences—RaTG13 is the closest relative of 2019-nCoV and they form a distinct lineage from other SARSr-CoVs (Fig. 1d and Extended Data Fig. 2). The receptor-binding spike protein encoded by the S gene was highly divergent from other CoVs (Extended Data Fig. 2), with less than 75% nucleotide sequence identity to all previously described SARSr-CoVs, except for a 93.1% nucleotide identity to RaTG13 (Extended Data Table 3). The S genes of 2019-nCoV and RaTG13 are longer than other SARSr-CoVs. The major differences in the sequence of the S gene of 2019-nCoV are the three short insertions in the N-terminal domain as well as changes in four out of five of the key residues in the receptor-binding motif compared with the sequence of SARS-CoV (Extended Data Fig. 3). Whether the insertions in the N-terminal domain of the S protein of 2019-nCoV confer sialic-acid-binding activity as it does in MERS-CoV needs to be further studied. The close phylogenetic relationship to RaTG13 provides evidence that 2019-nCoV may have originated in bats.
We rapidly developed a qPCR-based detection method on the basis of the sequence of the receptor-binding domain of the S gene, which was the most variable region of the genome (Fig. 1c). Our data show that the primers could differentiate 2019-nCoV from all other human coronaviruses including bat SARSr-CoV WIV1, which shares 95% identity with SARS-CoV (Extended Data Fig. 4a, b). Of the samples obtained from the seven patients, we found that six BALF and five oral swab samples were positive for 2019-nCoV during the first sampling, as assessed by qPCR and conventional PCR. However, we could no longer detect virus-positive samples in oral swabs, anal swabs and blood samples taken from these patients during the second sampling (Fig. 2a). However, we recommend that other qPCR targets, including the RdRp or envelope (E) genes are used for the routine detection of 2019-nCoV. On the basis of these findings, we propose that the disease could be transmitted by airborne transmission, although we cannot rule out other possible routes of transmission, as further investigation, including more patients, is required.
Fig. 2: Molecular and serological investigation of patient samples.
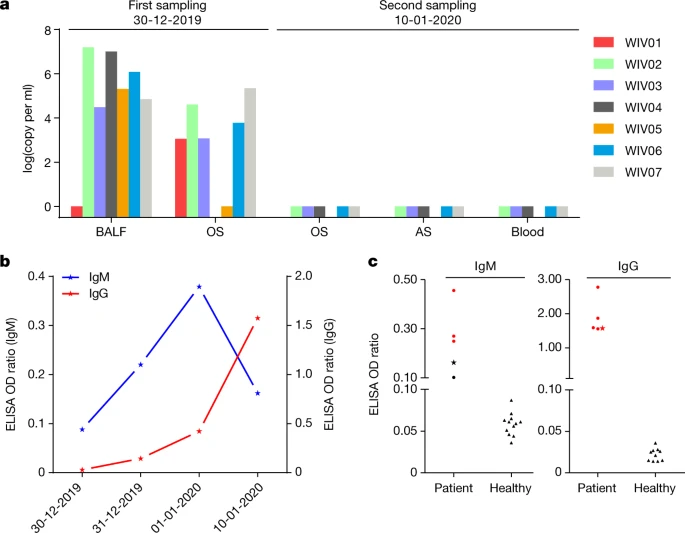
a, Molecular detection of 2019-nCoV in seven patients. Patient information can be found in Extended Data Tables 1, 2. Detection methods are described in the Methods. AS, anal swab; OS, oral swab. b, Dynamics of 2019-nCoV antibody levels in one patient who showed signs of disease on 23 December 2019 (ICU-06). OD ratio, optical density at 450–630 nm. The right and left y axes indicate ELISA OD ratios for IgM and IgG, respectively. c, Serological test of 2019-nCoV antibodies in five patients (Extended Data Table 2). The asterisk indicates data collected from patient ICU-06 on 10 January 2020. b, c, The cut-off was to 0.2 for the IgM analysis and to 0.3 for the IgG analysis, according to the levels of healthy controls.
For serological detection of 2019-nCoV, we used a previously developed nucleocapsid (N) protein from bat SARSr-CoV Rp3 as antigen for IgG and IgM enzyme-linked immunosorbent assays (ELISAs), as this protein shared 92% amino acid identity to N protein of 2019-nCoV (Extended Data Fig. 5) and showed no cross-reactivity against other human coronaviruses except SARSr-CoV7. We were only able to obtain five serum samples from the seven patients with viral infections. We monitored viral antibody levels in one patient (ICU-06) 7, 8, 9 and 18 days after the onset of disease (Extended Data Table 2). A clear trend was observed in the IgG and IgM titres, which increased over time, except that the IgM titre was decreased in the last sample (Fig. 2b). As a second analysis, we tested samples from 5 of the 7 virus-positive patients around 20 days after disease onset for the presence of viral antibodies (Extended Data Tables 1, 2). All patient samples—but not samples from healthy individuals—were strongly positive for viral IgG (Fig. 2b). There were also three IgM-positive samples, indicating an acute infection.
 ]
]