|
|
Post by Admin on Jan 27, 2020 19:36:04 GMT
The first people who contracted the deadly new coronavirus sweeping through Asia were infected when the virus jumped from animals to humans, and a new report points to the original animal source: bats. Scientists from the Wuhan Institute for Virology — located in China’s epicenter of the outbreak, now under quarantine — published a paper Thursday that confirmed the fast-spreading virus is in the same family as the severe acute respiratory syndrome (SARS) virus that hit Asia in 2003 and killed almost 800 people, and Middle East respiratory syndrome (MERS). Based on oral swabs, anal swabs, and blood collected from seven patients at Jinyintan Hospital in Wuhan, the scientists were able to genetically sequence the virus. Then they tested it against a database of known viruses and found a 96.2% match with a coronavirus known to live in bats, which is also believed to be the source of the SARS and MERS outbreaks. Ebola, the deadly virus that killed more than 11,000 people in West Africa between 2013 and 2016, is also believed to have originated from bats. The researchers found that the new virus uses the same receptors as SARS to hack into a victim’s lungs, causing symptoms such as coughing, headaches, and pneumonia. This paper is the first to describe the new virus, dubbed nCoV-2019, in detail. The outbreak is believed to have originated at a meat and seafood market in Wuhan where live animals were slaughtered and wild animals were also sold. Another report from China earlier this week claimed that the coronavirus may have originated in snakes, which were reportedly sold in the Wuhan market. However, that report also suggested that since snakes hunt bats in the wild, they could have been the original source of the virus. The new coronavirus, like SARS and MERS, is a zoonotic viral disease, meaning the first patients who were infected acquired these viruses directly from animals.  Since it emerged last month, the new virus has killed 26 people and infected 830, most of whom are in Wuhan, where authorities are trying to fast-track a new, 1000-bed hospital in just six days. The outbreak has come at the worst time for China as hundreds of millions of people will travel long distances this weekend to celebrate Lunar New Year, making the task of containing the spread of the virus much harder. In a bid to stop the virus spreading, authorities have imposed strict travel restrictions on 10 cities in the central province of Hubei, effectively locking down some 30 million people. But these efforts come after the virus has already spread to almost all areas of China, and internationally to Thailand, Hong Kong, Taiwan, Macau, Vietnam, South Korea, Japan, and the U.S., where a second confirmed case was reported in Chicago on Friday. Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin
23 Since the SARS outbreak 18 years ago, a large number of severe acute
24 respiratory syndrome related coronaviruses (SARSr-CoV) have been discovered
25 in their natural reservoir host, bats1-4. Previous studies indicated that some of
26 those bat SARSr-CoVs have the potential to infect humans5-7. Here we report the
27 identification and characterization of a novel coronavirus (nCoV-2019) which
28 caused an epidemic of acute respiratory syndrome in humans, in Wuhan, China.
29 The epidemic, started from December 12th, 2019, has caused 198 laboratory
30 confirmed infections with three fatal cases by January 20th 2020. Full-length
31 genome sequences were obtained from five patients at the early stage of the
32 outbreak. They are almost identical to each other and share 79.5% sequence
33 identify to SARS-CoV. Furthermore, it was found that nCoV-2019 is 96%
34 identical at the whole genome level to a bat coronavirus. The pairwise protein
35 sequence analysis of seven conserved non-structural proteins show that this virus
36 belongs to the species of SARSr-CoV. The nCoV-2019 virus was then isolated
37 from the bronchoalveolar lavage fluid of a critically ill patient, which can be
38 neutralized by sera from several patients. Importantly, we have confirmed that
39 this novel CoV uses the same cell entry receptor, ACE2, as SARS-CoV.
|
|
|
|
Post by Admin on Jan 27, 2020 21:45:08 GMT

41 Coronavirus has caused two large-scale pandemic in the last two decades, SARS and
42 MERS (Middle East respiratory syndrome)8,9. It was generally believed that SARSr
43 CoV, mainly found in bats, might cause future disease outbreak10. Here we report
44 on a series of unidentified pneumonia disease outbreaks in Wuhan, Hubei province,
45 central China (Extended Data Figure 1). Started from a local fresh seafood market, the
46 epidemic has resulted in 198 laboratory confirmed cases with three death according to
authorities so far12 47. Typical clinical symptoms of these patients are fever, dry cough,
48 dyspnea, headache, and pneumonia. Disease onset may result in progressive
49 respiratory failure due to alveolar damage and even death. The disease was
50 determined as viral induced pneumonia by clinicians according to clinical symptoms
51 and other criteria including body temperature rising, lymphocytes and white blood
52 cells decreasing (sometimes normal for the later), new pulmonary infiltrates on chest
53 radiography, and no obvious improvement upon three days antibiotics treatment. It
54 appears most of the early cases had contact history with the original seafood market,
55 and no large scale of human-to-human transmission was observed so far.
56
57 Samples from seven patients with severe pneumonia (six are seafood market peddlers
58 or delivers), who were enrolled in intensive unit cares at the beginning of the outbreak,
59 were sent to WIV laboratory for pathogen diagnosis (Extended Data Table 1). As a
60 CoV lab, we first used pan-CoV PCR primers to test these samples13, considering the
61 outbreak happened in winter and in a market, same environment as SARS. We found
62 five PCR positive. A sample (WIV04) collected from bronchoalveolar lavage fluid
63 (BALF) was analysed by metagenomics analysis using next-generation sequencing
64 (NGS) to identify potential etiological agents. Of the 1582 total reads obtained after
65 human genome filtering, 1378 (87.1%) matched sequences of SARSr-CoV (Fig. 1a).
66 By de novo assembly and targeted PCR, we obtained a 29,891-bp CoV genome that
67 shared 79.5% sequence identity to SARS-CoV BJ01 (GenBank accession number
68 AY278488.2). This sequence has been submitted to GISAID (accession no.
69 EPI_ISL_402124). Following the name by WHO, we tentatively call it novel
70 coronavirus 2019 (nCoV-2019). Four more full-length genome sequences of nCoV
71 2019 (WIV02, WIV05, WIV06, and WIV07) (GISAID accession nos.
72 EPI_ISL_402127-402130) that were above 99.9% identical to each other were
73 subsequently obtained from other four patients (Extended Data Table 2).
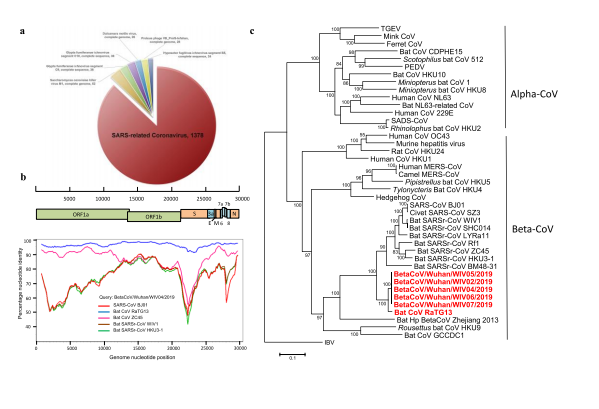 Fig. 1 | Genome characterization of nCoV-2019.
75 The virus genome consists of six major open reading frames (ORFs) common to
76 coronaviruses and a number of other accessory genes (Fig. 1b). Further analysis
77 indicates that some of the nCoV-2019 genes shared less than 80% nt sequence
78 identity to SARS-CoV. However, the seven conserved replicase domains in ORF1ab
79 that were used for CoV species classification, are 94.6% aa sequence identical
80 between nCoV-2019 and SARS-CoV, implying the two belong to same species
81 (Extended Data Table 3).
82
83 We then found a short RdRp region from a bat coronavirus termed BatCoV RaTG13
84 which we previously detected in Rhinolophus affinis from Yunnan Province showed
85 high sequence identity to nCoV-2019. We did full-length sequencing to this RNA
86 sample. Simplot analysis showed that nCoV-2019 was highly similar throughout the
87 genome to RaTG13 (Fig. 1c), with 96.2% overall genome sequence identity. The
88 phylogenetic analysis also showed that RaTG13 is the closest relative of the nCoV
89 2019 and form a distinct lineage from other SARSr-CoVs (Fig. 1d). The receptor
90 binding protein spike (S) gene was highly divergent to other CoVs (Extended Data
91 Figure 2), with less than 75% nt sequence identity to all previously described SARSr
92 CoVs except a 93.1% nt identity to RaTG13 (Extended Data Table 3). The S genes of
93 nCoV-2019 and RaTG13 S gene are longer than other SARSr-CoVs. The major
94 differences in nCoV-2019 are the three short insertions in the N-terminal domain, and
95 four out of five key residues changes in the receptor-binding motif, in comparison
96 with SARS-CoV (Extended Data Figure 3). The close phylogenetic relationship to
97 RaTG13 provides evidence for a bat origin of nCoV-2019.
|
|
|
|
Post by Admin on Jan 28, 2020 5:42:00 GMT
99 We rapidly developed a qPCR detection based on the receptor-binding domain of
100 spike gene, the most variable region among genome (Fig. 1c). Our data show the
101 primers could differentiate nCoV-2019 with all other human coronaviruses including
102 bat SARSr-CoV WIV1, which is 95% identity to SARS-CoV (Extended Data Figure
103 4a and 4b). From the seven patients, we found nCoV-2019 positive in six BALF and
104 five oral swab samples during the first sampling by qPCR and conventional PCR
105 (Extended Data Figure 4c). However, we can no longer find viral positive in oral
106 swabs, anal swabs, and blood from these patients during the second sampling (Fig.
107 2a). Based on these findings, we conclude that the disease should be transmitted
108 through airway, yet we can’t rule out other possibilities if the investigation extended
109 to include more patients.
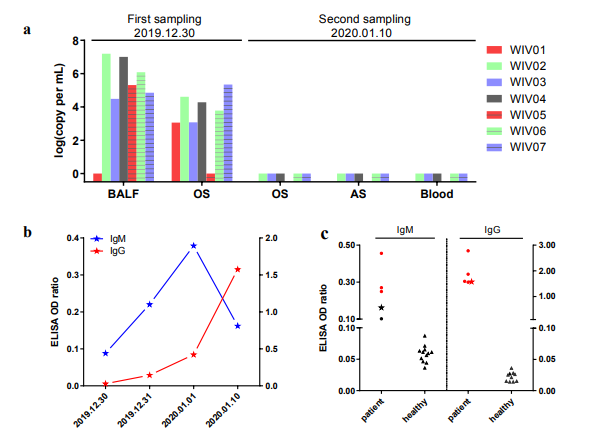 Fig. 2 | Molecular and serological investigation of patient samples.
111 For serological detection of nCoV-2019, we used previously developed bat SARSr
112 CoV Rp3 nucleocapsid protein (NP) as antigen in IgG and IgM ELISA test, which
113 showed no cross-reactivity against other human coronaviruses except SARSr-CoV7.
114 As a research lab, we were only able to get five serum samples from the seven viral
115 infected patients. We monitored viral antibody levels in one patient (ICU-06) at seven,
116 eight, nine, and eighteen days after disease onset (Extended Data Table 2). A clear
117 trend of IgG and IgM titre (decreased at the last day) increase was observed (Fig. 2b).
118 For a second investigation, we tested viral antibody for five of the seven viral positive
119 patients around twenty days after disease onset (Extended Data Table 1 and 2). All
120 patient samples, but not samples from healthy people, showed strong viral IgG
121 positive (Fig. 2b). We also found three IgM positive, indicating acute infection.
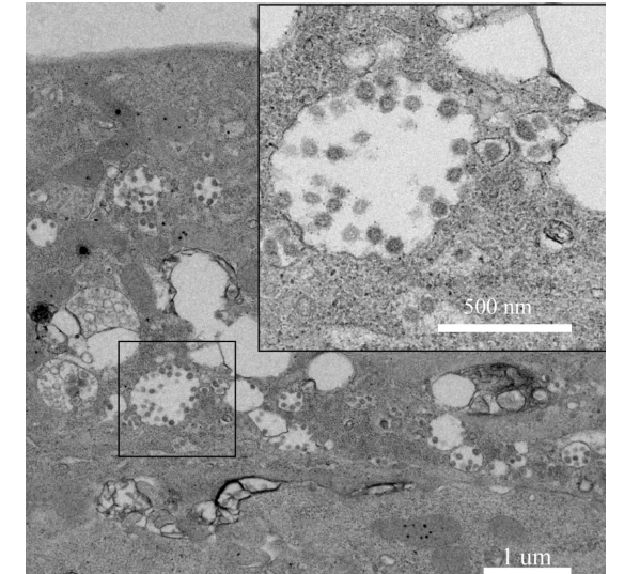 Fig. 3 | Virions. a, viral particles in the ultrathin sections under electron microscope at 200 kV, sample from viral infected Vero E6 cells
123 We then successfully isolated the virus (named nCoV-2019
124 BetaCoV/Wuhan/WIV04/2019), in Vero and Huh7 cells using BALF sample from
125 ICU-06 patient. Clear cytopathogenic effects were observed in cells after three days
126 incubation (Extended Data Figure 5a and 5b). The identity of the strain WIV04 was
127 verified in Vero E6 cells by immunofluorescence microscopy using cross-reactive
128 viral NP antibody (Extended Data Figure 5c and 5d), and by metagenomic sequencing,
129 from which most of the reads mapped to nCoV-2019 (Extended Data Figure 5e and
130 5f). Viral partials in ultrathin sections of infected cells displayed typical coronavirus
131 morphology under electron microscopy (Fig. 3). To further confirm the neutralization
132 activity of the viral IgG positive samples, we conducted serum-neutralization assays
133 in Vero E6 cells using the five IgG positive patient sera. We demonstrate that all
134 samples were able to neutralize 120 TCID50 nCoV-2019 at a dilution of 1:40-1:80.
135 We also show that this virus could be cross-neutralized by horse anti-SARS-CoV
136 serum at dilutions 1:80, further confirming the relationship of the two viruses
137 (Extended Data Table 4).
|
|
|
|
Post by Admin on Jan 28, 2020 21:34:35 GMT
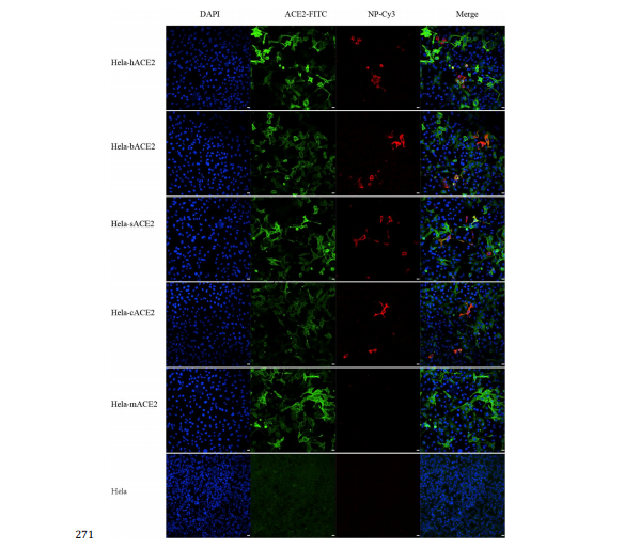 Fig. 4 | Analysis of nCoV-2019 receptor usage.
139 Angiotensin converting enzyme II (ACE2) was known as cell receptor for SARSCoV14
140 . To determine whether nCoV-2019 also use ACE2 as a cellular entry receptor,
141 we conducted virus infectivity studies using HeLa cells expressing or not expressing
142 ACE2 proteins from humans, Chinese horseshoe bats, civet, pig, and mouse. We
143 show that nCoV-2019 is able to use all but mouse ACE2 as an entry receptor in the
144 ACE2-expressing cells, but not cells without ACE2, indicating which is likely the cell
145 receptor of nCoV-2019 (Fig. 4). We also proved that nCoV-2019 does not use other
146 coronavirus receptors, aminopeptidase N and dipeptidyl peptidase 4 (Extended Data
147 Figure 6).
148
149 The study provides the first detailed report on nCoV-2019, the likely etiology agent
150 responsible for ongoing acute respiratory syndrome epidemic in Wuhan, central China.
151 Viral specific nucleotide positive and viral protein seroconversion observed in all
152 patients tested provides evidence of an association between the disease and the
153 presence of this virus. However, there are still many urgent questions to be answered.
154 We need more clinical data and samples to confirm if this virus is indeed the etiology
155 agent for this epidemic. In addition, we still don’t know if this virus continue evolving
156 and become more transmissible between human-to-human. Moreover, we don’t know
157 the transmission routine of this virus among hosts yet. We showed viral positive in
158 oral swabs, implying nCoV-2019 may be transmitted through airway. However, this
159 needs to be confirmed by extending detection range. Finally, based on our results, it
160 should be expected and worth to test if ACE2 targeting or SARS-CoV targeting drugs
161 can be used for nCoV-2019 patients. At this stage, we know very little about the virus,
162 including basic biology, animal source or any specific treatment. The almost identical
163 sequences of this virus in different patients imply a probably recent introduction in
164 humans, thus future surveillance on viral mutation and transmission ability and
165 further global research attention are urgently needed.
166
167 ACKNOWLEDGEMENTS: We thank the Pei Zhang and An-na Du from WIV core
168 facility and technical support for their help with producing TEM micrographs. This
169 work was jointly supported by the Strategic Priority Research Program of the Chinese
|
|
|
|
Post by Admin on Jan 29, 2020 21:50:57 GMT
A new genetic analysis of 10 genome sequences of novel coronavirus (2019-nCoV) from nine patients in Wuhan finds that the virus is most closely related to two bat-derived SARS-like coronaviruses, according to a study published in The Lancet: www.biorxiv.org/content/10.1101/2020.01.22.914952v1 The authors say that although their analysis suggests that bats might be the original host of the virus, an animal sold at the Huanan seafood market in Wuhan might represent an intermediate host that enables the emergence of the virus in humans. For this reason, the future evolution, adaptation and spread of this virus requires urgent investigation. In the study, the authors report the epidemiological data of nine patients who were diagnosed with viral pneumonia of unidentified cause. Cell and secretion samples were taken from the patients' lungs to harvest samples of the 2019-nCoV virus, which were analysed to determine the origin of the virus and how it enters human cells. Eight of the patients had visited the Huanan seafood market. One patient had never visited the market, but had stayed in a hotel near the market before the onset of their illness. The authors found 2019-nCoV in all 10 genetic samples taken from the patients - including eight complete genomes, and two partial genomes. The genetic sequences of the samples were nearly identical (shared more than 99.98% of the same genetic sequence) - which indicates a very recent emergence of the virus into humans.  "It is striking that the sequences of 2019-nCoV described here from different patients were almost identical. This finding suggests that 2019-nCoV originated from one source within a very short period and was detected relatively rapidly. However, as the virus transmits to more individuals, constant surveillance of mutations arising is needed," says one of lead authors Professor Weifeng Shi, Key Laboratory of Etiology and Epidemiology of Emerging Infectious Diseases in Universities of Shandong, Shandong First Medical University and Shandong Academy of Medical Sciences, China. Comparing the 2019-nCoV genetic sequence with a library of viruses, the authors found that the most closely related viruses were two SARS-like coronaviruses of bat origin - bat-SL-CoVZC45 and bat-SL-CoVZXC21 - which shared 88% of the genetic sequence. 2019-nCoV was more genetically distant to the human SARS virus (which shared about 79% of the genetic sequence) and the Middle East respiratory syndrome (MERS) virus (which shared about 50% of the genetic sequence). Studying the spike protein of the virus (how it binds then enters human cells), the authors found that 2019-nCoV and human SARS virus have similar structures, despite some small differences. As a result, the authors suggest that 2019-nCoV might use the same molecular doorway to enter the cells as SARS (a receptor called ACE2), but note that this will require confirmation.  Based on their data, the authors say that it seems likely that the 2019-nCoV causing the Wuhan outbreak might also be initially hosted by bats and transmitted to humans via a currently unknown wild animal sold at the Huanan seafood market. They say that it is more likely that bat coronaviruses are mutating, than 2019-nCoV - meaning that 2019-nCoV is unlikely to have emerged due to a chance mutation. However, more information is needed, and if a more closely related animal virus is identified, this suggestion could be wrong. "These data are consistent with a bat reservoir for coronaviruses in general and 2019-nCoV in particular. However, despite the importance of bats, it seems likely that another animal host is acting as an intermediate host between bats and humans," says Professor Guizhen Wu, Chinese Center for Disease Control and Prevention. Explaining this she notes: "First, the outbreak was first reported in late December, 2019, when most bat species in Wuhan are hibernating. Second, no bats were sold or found at the Huanan seafood market, whereas many non-aquatic animals (including mammals) were. Third, the similarities in the genetic sequences between 2019-nCoV and its close relatives bat-SL-CoVZC45 and bat-SL-CoVZXC21 were less than 90%, meaningthese two bat-derived coronaviruses are not direct ancestors of 2019-nCoV. Fourth, in both SARS and MERS, bats acted as the natural reservoir, with another animal acting as an intermediate host, and with humans as terminal hosts.This again highlights the hidden virus reservoir in wild animals and their potential to spill over into human populations." |
|

