INTRODUCTION
The coronavirus disease 2019 (COVID-19) epidemic started in late December 2019 in Wuhan,
the capital of Central China's Hubei Province. Since then, it has rapidly spread across China
and in other countries, raising major global concerns. The etiological agent is a novel
coronavirus, SARS-CoV-2, named for the similarity of its symptoms to those induced by the
severe acute respiratory syndrome. As of February 28, 2020, 78,959 cases of SARS-CoV-2
infection have been confirmed in China, with 2,791 deaths. Worryingly, there have also been
more than 3,664 confirmed cases outside of China in 46 countries and areas
(https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/),
raising significant doubts about the likelihood of successful containment. Further, the
genomic sequences of SARS-CoV-2 viruses isolated from a number of patients share
sequence identity higher than 99.9%, suggesting a very recent host shift into humans [1-3].
Coronaviruses are naturally hosted and evolutionarily shaped by bats [4, 5]. Indeed, it has
been postulated that most of the coronaviruses in humans are derived from the bat reservoir [6,
7]. Unsurprisingly, several teams have recently confirmed the genetic similarity between
SARS-CoV-2 and a bat betacoronavirus of the sub-genus Sarbecovirus [8-13]. The
whole-genome sequence identity of the novel virus has 96.2% similarity to a bat
SARS-related coronavirus (SARSr-CoV; RaTG13) collected in Yunnan province, China [2,
14], but is not very similar to the genomes of SARS-CoV (about 79%) or MERS-CoV (about
50%) [1, 15]. It has also been confirmed that the SARS-CoV-2 uses the same receptor, the
angiotensin converting enzyme II (ACE2), as the SARS-CoV [11]. Although the specific
route of transmission from natural reservoirs to humans remains unclear [5, 13], several
studies have shown that pangolins may have provided a partial spike gene to SARS-CoV-2;
the critical functional sites in the spike protein of SAR-CoV-2 are nearly identical to one
identified in a virus isolated from a pangolin [16-18].
Despite these recent discoveries, several fundamental issues related to the evolutionary
patterns and driving forces behind this outbreak of SARS-CoV-2 remain unexplored [19].
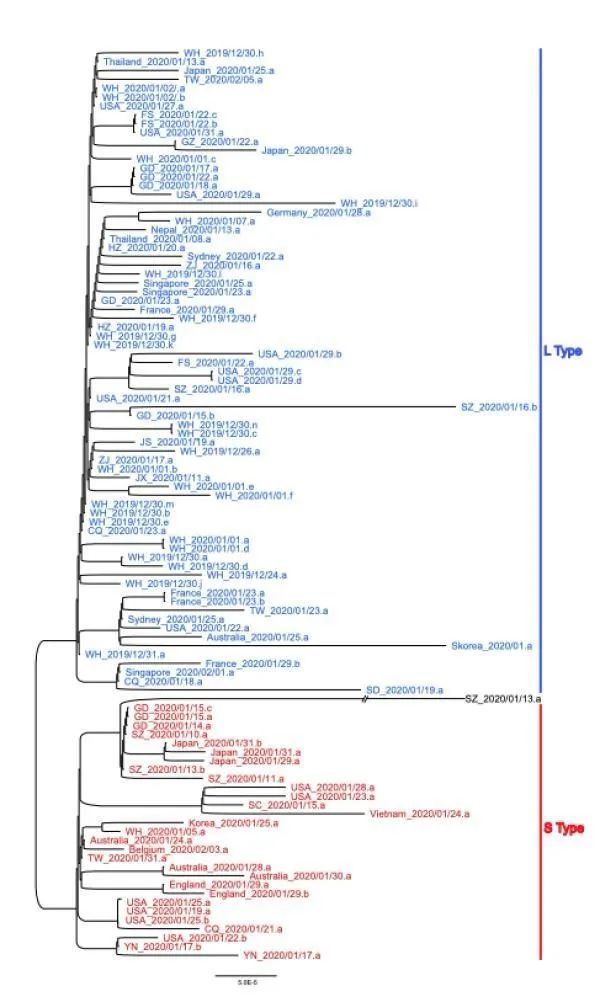
Herein, we investigated the extent of molecular divergence between SARS-CoV-2 and other
related coronaviruses and carried out population genetic analyses of 103 sequenced genomes
of SARS-CoV-2. This work provides new insights into the factors driving the evolution of
SARS-CoV-2 and its pattern of spread through the human population.
RESULTS
Molecular phylogeny and divergence between SARS-CoV-2 and related coronaviruses.
For each annotated ORF in the reference genome of SARS-CoV-2 (NC_045512), we
extracted the orthologous sequences in human SARS-CoV, four bat
SARS-related coronaviruses (SARSr-CoV: RaTG13, ZXC21, ZC45, and BM48-31), one
Pangolin SARSr-CoV from Guangdong (GD) [17], and six Pangolin SARSr-CoV genomes
from Guangxi (GX) [18] (Table S1). We aligned the coding sequences (CDSs) based on the
protein alignments (see Materials and Methods). Most ORFs annotated from SARS-CoV-2
were found to be conserved in other viruses, except for ORF8 and ORF10 (Table 1). The
protein sequence of SARS-CoV-2 ORF8 shared very low similarity with sequences in
SARS-CoV and BM48-31, and ORF10 had a premature stop codon in both SARS-CoV and
BM48-31 (Fig. S1). A one-base deletion caused a frame-shift mutation in ORF10 of ZXC21
(Fig. S1).
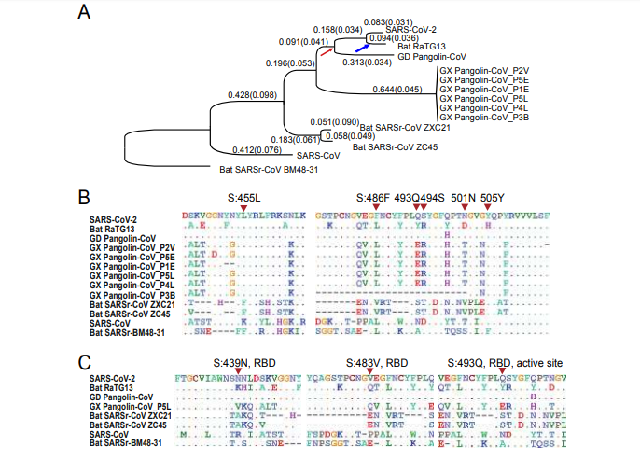
Figure 1. Molecular divergence and selective pressures during the evolution of
SARS-CoV-2 and related viruses.
A. The phylogenetic tree of SARS-CoV-2 and the related Coronaviruses. The branch length
(dS) is presented, and the dN/dS (ω) value is given in the parenthesis. The phylogenetic tree
was reconstructed with the synonymous sites in the concatenated CDSs of nine conserved
ORFs (orf1ab, E, M, N, S, ORF3a, ORF6, ORF7a and ORF7b).
B. Conservation of 6 critical amino acid residues in the spike (S) protein. The critical active
sites are Y442, L472, N479, D480, T487, and Y491 in SARS-CoV, and they correspond to
L455, F486, Q493, S494, N501, and Y505 in SARS-CoV-2 (marked with inverted triangles),
respectively.
C. Three candidate positively selected sites (marked with inverted triangles) in the
receptor-binding domain (RBD) of spike protein (S:439N, S:483V and S:493Q) and the
surrounding 10 amino acids
Notably, the dS value varied considerably across genes in SARS-CoV-2 and the other viruses
analyzed. In particular, the spike gene (S) consistently exhibited larger dS values than other
genes (Table 1). This pattern became clear when we calculated the dS value for each branch
in Fig. 1A for the spike gene versus the concatenated sequences of the remaining genes (Fig.
S2). In each branch, the dS of spike was 2.22 ± 1.35 (mean ± SD) times as large as that of the
other genes. This extremely elevated dS value of spike could be caused either by a high
mutation rate or by natural selection that favors synonymous substitutions. Synonymous
substitutions may serve as another layer of genetic regulation, guiding the efficiency of
mRNA translation by changing codon usage [21]. If positive selection is the driving force for
the higher synonymous substation rate seen in spike, we expect the frequency of optimal
codons (FOP) of spike to be different from that of other genes. However, our codon usage
bias analysis (Table S2) suggests the FOP of spike was only slightly higher than that of the
genomic average (0.717 versus 0.698, see Materials and Methods). Thus, we believe that the
elevated synonymous substitution rate measured in spike is more likely caused by higher
mutational rates; however, the underlying molecular mechanism remains unclear.
Both SARS-CoV and SARS-CoV-2 bind to ACE2 through the RBD of spike protein in order
to initiate membrane fusion and enter human cells [1, 2, 22-26]. Five out of the six critical
amino acid (AA) residues in RBD were different between SARS-CoV-2 and SARS-CoV (Fig.
1B), and a 3D structural analysis indicated that the spike of SARS-CoV-2 has a higher
binding affinity to ACE2 than SARS-CoV [23]. Intriguingly, these same six critical AAs are
identical between GD Pangolin-CoV and SARS-CoV-2 [16]. In contrast, although the
genomes of SARS-CoV-2 and RaTG13 are more similar overall, only one out of the six
functional sites are identical between the two viruses (Fig. 1B). It has been proposed that the
SARS-CoV-2 RBD region of the spike protein might have resulted from recent recombination
events in pangolins [16-18]. Although several ancient recombination events have been
described in spike [27, 28], it also seems likely that the identical functional sites in
SARS-CoV-2 and GD Pangolin-CoV may actually the result of coincidental convergent
evolution [18].
If the functional AA residues in the SARS-CoV-2 RBD region were acquired from GD
Pangolin-CoV in a very recent recombination event, we would expect the nucleotide
sequences of this region to be nearly identical between the two viruses. However, for the CDS
sequences that span five critical AA sites in the SARS-CoV-2 spike (ranging from codon 484
to 507, covering five adjacent functional sites: F486, Q493, S494, N501, and Y505; Fig. S3),
we estimated dS = 0.411, dN = 0.019, and ω= 0.046 between SARS-CoV-2 and GD
Pangolin-CoV. By assuming the synonymous substitution rate (u) of 1.67-4.67 x 10-3
/site/year, as estimated in SARS-CoV [29], the recombination/introgression, if it occurred at all,
would be estimated to happen approximately 19.8-55.4 years ago. Here, the formula
was used to calculate divergence time; note that the increased mutational rate of
spike was considered for this calculation. Thus, it seems very unlikely that SARS-CoV-2
originated from the GD Pangolin-CoV due to a very recent recombination event.
Alternatively, it seems more likely that a high mutation rate in spike, coupled with strong
natural selection, has shaped the identical functional AA residues between these two viruses,
as proposed previously [18]. Although these sites are maintained in SARS-CoV-2 and GD
To investigate the phylogenetic relationships between these viruses at the genomic scale, we
concatenated coding regions (CDSs) of the nine conserved ORFs (orf1ab, E, M, N, S, ORF3a,
ORF6, ORF7a, and ORF7b) and reconstructed the phylogenetic tree using the synonymous
sites (Fig. 1A). We also used CODEML in the PAML [20] to infer the ancestral sequence of
each node and calculated the dN (nonsynonymous substitutions per nonsynonymous site), dS
(synonymous substitutions per synonymous site), and dN/dS (ω) values for each branch (Fig.
1A). In parallel, we also calculated the pairwise dN, dS, and ω values between SARS-CoV-2
and another virus (Table 1).
The genome-wide phylogenetic tree indicated that SARS-CoV-2 was closest to RaTG13,
followed by GD Pangolin SARSr-CoV, then by GX Pangolin SARSr-CoVs, then by ZC45
and ZXC21, then by human SARS-CoV, and finally by BM48-31(Fig. 1A). Notably, we
found that the nucleotide divergence at synonymous sites between SARS-CoV-2 and other
viruses was much higher than previously anticipated. For example, although the overall
genomic nucleotides overall differ ~4% between SARS-CoV-2 and RaTG13, the genomic
average dS was 0.17, which means the divergence at the neutral sites is 17% between these
two viruses (Table 1). This is because the nonsynonymous sites are usually under stronger
negative selection than synonymous sites, and calculating sequence differences without
separating these two classes of sites may underestimate the extent of molecular divergence by
several folds.
Pangolin-CoV, mutations may have changed the residues in the RaTG13 lineage after it
diverged from SARS-CoV-2 (the blue arrow in Fig. 1A). In summary, it seems that the shared
identity of critical AA sites between SARS-CoV-2 and GD Pangolin-CoV might be due to
random mutations coupled with natural selection, and not necessarily recombination.
Selective constraints and positive selection during the evolution of SARS-CoV-2 and
related coronaviruses
The genome-wide ω value between SARS-CoV-2 and other viruses ranged from 0.044 to
0.124 (Table 1), indicative of strong negative selection on the nonsynonymous sites. In other
words, 87.6% to 95.6% of the nonsynonymous mutations were removed by negative selection
during viral evolution. To determine the extent of positive selection, we concatenated the
CDS sequences of 9 conserved ORFs in all the viruses in Fig. 1A and fitted the M7 (beta:
neutral and negative selection) and M8 (beta + ω>1:neutral, negative selection, and positive
selection) model using CODEML (Materials and Methods). The M8 model (lnL =
-104,813.732, np =18) was a significantly better fit than the M7 (lnL = -105,063.284, np = 16)
model (P < 10-10), suggesting that some AA substitutions were favored by positive Darwinian
selection (but not necessarily in the SARS-CoV-2 lineage).Under the M8 model, 98.48% (p0)
of the nonsynonymous substitutions were estimated under neutral evolution or purifying
selection (0⩽ω⩽1), and 1.52% (p1) of the nonsynonymous substitutions were under positive
selection (ω = 1.50). A Bayes Empirical Bayes (BEB) analysis suggested that 10 AA sites
showed strong signals of positive selection, and, interestingly, three of those were located in
the RBD of spike, including at one critical site (Fig. 1C and Fig. S4). Thus, although these
coronaviruses were generally under very strong negative selection, positive selection was also
responsible for the evolution of protein sequences. The putatively positively-selected sites
might serve as candidates for further functional studies.