Post by Admin on Aug 9, 2020 19:21:53 GMT
Discussion
In this article, we present a new method, called ARGweaver-D, for sampling ancestral recombination graphs (ARGs) under an arbitrary demographic model, and show that this method is particularly powerful for identifying introgressed regions. Like our previous ARGweaver method, ARGweaver-D is limited to use with relatively small sample sizes (up to about 100 haploid genomes) owing to its considerable computational demands, but even in this setting it shows excellent power in the detection of introgression, particularly older events. In addition, ARGweaver-D has several other benefits over alternative methods for detection of gene flow. For example, it does not require a reference panel of non-introgressed individuals, and it can simultaneously identify introgression stemming from multiple migration events, as well as from both sampled or unsampled populations. In addition, ARGweaver-D does not rely on summary statistics, but uses a model of coalescence and recombination to generate local gene trees that are most consistent with the observed patterns of variation, even for unphased genomes. By incorporating all this information simultaneously, it can successfully distinguish migration from incomplete lineage sorting, and tease apart different migration events that produce similar D statistics (such as Sup→Den and Hum→Nea). The ARGweaver-D code is freely available and can be applied to any similar collection of DNA sequences and user-defined demographic scenario.
While we have focused in this article on introgression, ARGweaver-D is a fully general method for demography-aware ARG inference and could potentially have many other applications. The ARGs sampled by ARGweaver-D—by reflecting constraints on coalescence consistent with isolation and migration scenarios, as well as differences in effective size across populations—are likely to be considerably more accurate than those inferred by ARGweaver, which assumes a panmictic population of constant size. These differences may be particularly important in cases in which populations have dramatically different sizes (as with modern and archaic humans) or in cases of complete or near-complete population isolation. These improvements could be relevant in many applications of ARG inference, including the detection of sequences under selection or estimation of allele ages [21]. Moreover, ARGweaver-D can be useful in evaluating the relative likelihoods of alternative demographic models (although formal model testing in this framework remains a challenging and unsolved problem). Finally, it is worth emphasizing that in multi-population settings with limited migration, ARGweaver-D can be considerably more efficient than ARGweaver, because constraints on permissible coalescence events result in a substantial reduction in the state space of the HMM used for the core “threading” operation.
A recently published method, dical-admix [28], is similar to ARGweaver-D in that it is designed to accommodate generic demographic models and considers the full haplotype structure of the input sequences. This method appears to be quite powerful, but it does assume that there are only a few admixed individuals, and that other genomes are “trunk” lineages that help define the haplotype structure of their respective populations. Therefore, unlike ARGweaver-D, dical-admix cannot infer admixture from an unsampled population, nor is it designed to work when all individuals have some degree of admixed ancestry. Additionally, ARGweaver-D has the advantage of allowing for unphased genomes, which is important since there are currently too few Neanderthal or Denisovan samples to permit reliable phasing. In addition, two methods have recently been published that permit approximate gene-tree or ARG inference on much larger scales (with up to hundreds of thousands of samples) [29, 30] but, as yet, these methods do not consider the constraints of a demographic model or allow for uncertainty in the ARG given the data. More work would be needed to optimize them for use in detecting introgression events.
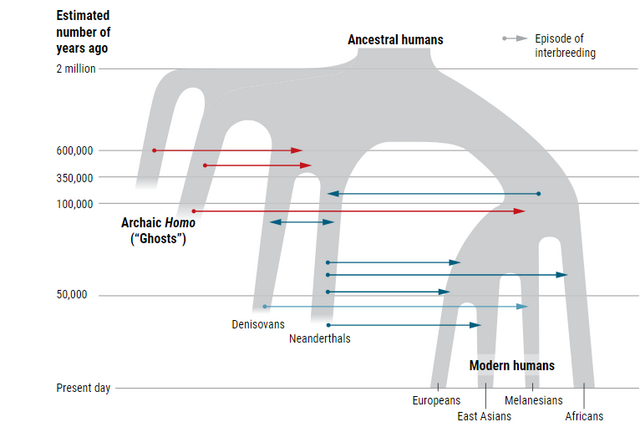
By applying ARGweaver-D to modern and archaic hominins, we confirmed that a substantial proportion of the Neanderthal genome consists of regions introgressed from ancient humans. While we identified only 3% of the Neanderthal genome as introgressed, a rough extrapolation based on our estimated rates of true and false positives suggests that the true fraction is ∼7% (S1 Text). Thus, the Neanderthal genome likely includes a larger contribution from ancient humans than modern non-African human genomes include from Neanderthal introgression.
Our follow-up analysis based on the frequencies of introgressed elements among the two diploid Neanderthal genomes suggests that the Hum→Nea gene flow occurred roughly between 200 and 300kya, within the limits of accuracy imposed by our assumed demographic model, mutation rates, and generation time. As previously noted [8, 9], because contact between modern humans and Neanderthals most likely took place in Eurasia, this timeline appears to be inconsistent with a genetic exchange involving the direct ancestors of most present-day Eurasians, who migrated out of Africa ∼50kya. Instead, our timeline suggests an earlier migration, occurring at least 200kya. Notably, orthogonal lines of evidence now support the possibility of one or more such early migrations out of Africa. For example, mitochondrial DNA from Neanderthals has much lower divergence than expected from human mtDNA (with the exception of the Sima de los Huesos samples, which cluster with Denisovans) [31]. This observation suggests that most Neanderthal mtDNA is introgressed from ancient humans. A recent study [32] bounded this introgression event at ≥ 270kya, in rough agreement with our estimated timeline, under the assumption that the recently analyzed Hohlenstein-Stadel sample and all sequenced Late Pleistocene Neanderthals share a common mtDNA origin outside of Africa [31]. Beyond this genetic evidence, there have also been discoveries of ancient fossils with human characteristics outside of Africa, including a 180kya jawbone from Misliya Cave in Israel [33], and a 210kya fossil from Apidima Cave in southern Greece with human features [34]. These findings suggest that early modern humans were present on the Eurasian continent at roughly the time at which Hum→Nea gene flow is estimated to have occurred. These early migrating humans may later have gone extinct, leaving a genetic trace only in introgressed segments in Neanderthals.
Aside from the issue of timing is the question of the possible functional impact of the Hum→Nea introgression. This question has proved challenging due to the myriad ascertainment biases—known and unknown—that affect the power to detect introgressed regions. Even in the case of Nea→Hum migration, for which power to detect introgression is high, earlier claims of depletion near genes, as well as decreasing levels of introgression over time, have been recently called into question [13, 28]. The strongest remaining pieces of evidence for negative selection against Nea→Hum introgression are the depletion on the X chromosome and in several other genomic deserts. But for the Hum→Nea event, we see no depletion on the X, and while we had too few samples to detect deserts across Neanderthals, we did confirm that previously identified Nea→Hum deserts are not depleted for Hum→Nea introgression. We do see a slight decrease in Hum→Nea introgression on the X chromosome in the Vindija Neanderthal compared to the Altai, which could be explained by weak negative selection removing some introgressed regions in the ∼70ky interval between the ages of these fossils. An interesting question is whether the observed absence of negative selection reflects healthy variation introduced by human introgression into the Neanderthal genome, or a Neanderthal population that was too small for efficient removal of deleterious variants. With additional archaic samples, it may be possible to address these questions.
ARGweaver-D also identified 1% of the Denisovan genome as introgressed from a super-archaic hominin. Previous studies have attributed roughly 6% of the Denisovan genome to this event [9], but ours is the first study to identify specific introgressed regions. Our apparent weak power to detect these regions suggests that the introgressing population was not too highly diverged from other hominins; according to our simulations, the observed sensitivity is more consistent with a divergence time of 1Mya than 1.5Mya. Still, we identify 27Mb of putative super-archaic sequence from this previously unsequenced hominin, and we estimate that ∼15% of these regions have been passed on to at least one modern humans through Den→Hum introgression. Thus, our analysis suggests that at least about 4Mb of modern human genomes derives from an unknown but highly diverged archaic hominin, possibly Homo erectus, through at least two separate introgression events. Considering our lack of power, the true contribution could be as much as six times larger. It may be possible to identify more of this super-archaic sequence by applying ARGweaver-D to a recently sequenced set of 161 Oceanian genomes [35].
Several studies have suggested super-archaic introgression into various African populations [10, 11, 36]. However, ARGweaver-D only detected a low rate of Sup→Afr introgression, somewhat below our estimated false positive rate. Notably, however, the power to identify introgression from an unsequenced population is highly dependent on the population size of the recipient population: the larger the population, the deeper the coalescences are within that population, making it more difficult to discern which long branches might be explained by super-archaic introgression. In the case of Africans, we used a population size of 23,700, which was our best estimate from previous runs of G-PhoCS [8, 14]. If we had used a smaller population size, ARGweaver-D would have produced more Sup→Afr predictions, but possibly with a substantially higher false positive rate. In fact, one of the drawbacks of ARGweaver-D is that it depends on a demographic model, and choosing the wrong model can lead to spurious results. We have shown that ARGweaver-D is fairly robust to modest misspecifications in the model, but we recommend that careful demographic analysis be performed before running ARGweaver-D to ensure that the best possible model is used.
Finally, we detected small amounts of introgression for two additional events: Hum→Den and Sup→Nea. Because no previous evidence has been reported for these events, we expected that our predictions would be roughly in line with our false positive rates. Indeed, for the Hum→Den event, we predicted a slightly smaller fraction (0.37%) than the estimated false positive rate from simulations (0.41%). For Sup→Nea, however, we predicted 0.75% of the genome to be introgressed, which is slightly higher than the corresponding estimated false positive rate (0.65%). While these fractions are too small to draw strong conclusions, it is plausible that if Homo erectus mixed with the Denisovans, they may have also mixed with Neanderthals, perhaps in the Middle East; or perhaps DNA passed from Homo erectus to Neanderthal through the Denisovans. Altogether, given the number of gene flow events now documented among ancient hominins, it may be reasonable to assume that genetic exchange was likely whenever two groups overlapped in time and space.
In this article, we present a new method, called ARGweaver-D, for sampling ancestral recombination graphs (ARGs) under an arbitrary demographic model, and show that this method is particularly powerful for identifying introgressed regions. Like our previous ARGweaver method, ARGweaver-D is limited to use with relatively small sample sizes (up to about 100 haploid genomes) owing to its considerable computational demands, but even in this setting it shows excellent power in the detection of introgression, particularly older events. In addition, ARGweaver-D has several other benefits over alternative methods for detection of gene flow. For example, it does not require a reference panel of non-introgressed individuals, and it can simultaneously identify introgression stemming from multiple migration events, as well as from both sampled or unsampled populations. In addition, ARGweaver-D does not rely on summary statistics, but uses a model of coalescence and recombination to generate local gene trees that are most consistent with the observed patterns of variation, even for unphased genomes. By incorporating all this information simultaneously, it can successfully distinguish migration from incomplete lineage sorting, and tease apart different migration events that produce similar D statistics (such as Sup→Den and Hum→Nea). The ARGweaver-D code is freely available and can be applied to any similar collection of DNA sequences and user-defined demographic scenario.
While we have focused in this article on introgression, ARGweaver-D is a fully general method for demography-aware ARG inference and could potentially have many other applications. The ARGs sampled by ARGweaver-D—by reflecting constraints on coalescence consistent with isolation and migration scenarios, as well as differences in effective size across populations—are likely to be considerably more accurate than those inferred by ARGweaver, which assumes a panmictic population of constant size. These differences may be particularly important in cases in which populations have dramatically different sizes (as with modern and archaic humans) or in cases of complete or near-complete population isolation. These improvements could be relevant in many applications of ARG inference, including the detection of sequences under selection or estimation of allele ages [21]. Moreover, ARGweaver-D can be useful in evaluating the relative likelihoods of alternative demographic models (although formal model testing in this framework remains a challenging and unsolved problem). Finally, it is worth emphasizing that in multi-population settings with limited migration, ARGweaver-D can be considerably more efficient than ARGweaver, because constraints on permissible coalescence events result in a substantial reduction in the state space of the HMM used for the core “threading” operation.
A recently published method, dical-admix [28], is similar to ARGweaver-D in that it is designed to accommodate generic demographic models and considers the full haplotype structure of the input sequences. This method appears to be quite powerful, but it does assume that there are only a few admixed individuals, and that other genomes are “trunk” lineages that help define the haplotype structure of their respective populations. Therefore, unlike ARGweaver-D, dical-admix cannot infer admixture from an unsampled population, nor is it designed to work when all individuals have some degree of admixed ancestry. Additionally, ARGweaver-D has the advantage of allowing for unphased genomes, which is important since there are currently too few Neanderthal or Denisovan samples to permit reliable phasing. In addition, two methods have recently been published that permit approximate gene-tree or ARG inference on much larger scales (with up to hundreds of thousands of samples) [29, 30] but, as yet, these methods do not consider the constraints of a demographic model or allow for uncertainty in the ARG given the data. More work would be needed to optimize them for use in detecting introgression events.
By applying ARGweaver-D to modern and archaic hominins, we confirmed that a substantial proportion of the Neanderthal genome consists of regions introgressed from ancient humans. While we identified only 3% of the Neanderthal genome as introgressed, a rough extrapolation based on our estimated rates of true and false positives suggests that the true fraction is ∼7% (S1 Text). Thus, the Neanderthal genome likely includes a larger contribution from ancient humans than modern non-African human genomes include from Neanderthal introgression.
Our follow-up analysis based on the frequencies of introgressed elements among the two diploid Neanderthal genomes suggests that the Hum→Nea gene flow occurred roughly between 200 and 300kya, within the limits of accuracy imposed by our assumed demographic model, mutation rates, and generation time. As previously noted [8, 9], because contact between modern humans and Neanderthals most likely took place in Eurasia, this timeline appears to be inconsistent with a genetic exchange involving the direct ancestors of most present-day Eurasians, who migrated out of Africa ∼50kya. Instead, our timeline suggests an earlier migration, occurring at least 200kya. Notably, orthogonal lines of evidence now support the possibility of one or more such early migrations out of Africa. For example, mitochondrial DNA from Neanderthals has much lower divergence than expected from human mtDNA (with the exception of the Sima de los Huesos samples, which cluster with Denisovans) [31]. This observation suggests that most Neanderthal mtDNA is introgressed from ancient humans. A recent study [32] bounded this introgression event at ≥ 270kya, in rough agreement with our estimated timeline, under the assumption that the recently analyzed Hohlenstein-Stadel sample and all sequenced Late Pleistocene Neanderthals share a common mtDNA origin outside of Africa [31]. Beyond this genetic evidence, there have also been discoveries of ancient fossils with human characteristics outside of Africa, including a 180kya jawbone from Misliya Cave in Israel [33], and a 210kya fossil from Apidima Cave in southern Greece with human features [34]. These findings suggest that early modern humans were present on the Eurasian continent at roughly the time at which Hum→Nea gene flow is estimated to have occurred. These early migrating humans may later have gone extinct, leaving a genetic trace only in introgressed segments in Neanderthals.
Aside from the issue of timing is the question of the possible functional impact of the Hum→Nea introgression. This question has proved challenging due to the myriad ascertainment biases—known and unknown—that affect the power to detect introgressed regions. Even in the case of Nea→Hum migration, for which power to detect introgression is high, earlier claims of depletion near genes, as well as decreasing levels of introgression over time, have been recently called into question [13, 28]. The strongest remaining pieces of evidence for negative selection against Nea→Hum introgression are the depletion on the X chromosome and in several other genomic deserts. But for the Hum→Nea event, we see no depletion on the X, and while we had too few samples to detect deserts across Neanderthals, we did confirm that previously identified Nea→Hum deserts are not depleted for Hum→Nea introgression. We do see a slight decrease in Hum→Nea introgression on the X chromosome in the Vindija Neanderthal compared to the Altai, which could be explained by weak negative selection removing some introgressed regions in the ∼70ky interval between the ages of these fossils. An interesting question is whether the observed absence of negative selection reflects healthy variation introduced by human introgression into the Neanderthal genome, or a Neanderthal population that was too small for efficient removal of deleterious variants. With additional archaic samples, it may be possible to address these questions.
ARGweaver-D also identified 1% of the Denisovan genome as introgressed from a super-archaic hominin. Previous studies have attributed roughly 6% of the Denisovan genome to this event [9], but ours is the first study to identify specific introgressed regions. Our apparent weak power to detect these regions suggests that the introgressing population was not too highly diverged from other hominins; according to our simulations, the observed sensitivity is more consistent with a divergence time of 1Mya than 1.5Mya. Still, we identify 27Mb of putative super-archaic sequence from this previously unsequenced hominin, and we estimate that ∼15% of these regions have been passed on to at least one modern humans through Den→Hum introgression. Thus, our analysis suggests that at least about 4Mb of modern human genomes derives from an unknown but highly diverged archaic hominin, possibly Homo erectus, through at least two separate introgression events. Considering our lack of power, the true contribution could be as much as six times larger. It may be possible to identify more of this super-archaic sequence by applying ARGweaver-D to a recently sequenced set of 161 Oceanian genomes [35].
Several studies have suggested super-archaic introgression into various African populations [10, 11, 36]. However, ARGweaver-D only detected a low rate of Sup→Afr introgression, somewhat below our estimated false positive rate. Notably, however, the power to identify introgression from an unsequenced population is highly dependent on the population size of the recipient population: the larger the population, the deeper the coalescences are within that population, making it more difficult to discern which long branches might be explained by super-archaic introgression. In the case of Africans, we used a population size of 23,700, which was our best estimate from previous runs of G-PhoCS [8, 14]. If we had used a smaller population size, ARGweaver-D would have produced more Sup→Afr predictions, but possibly with a substantially higher false positive rate. In fact, one of the drawbacks of ARGweaver-D is that it depends on a demographic model, and choosing the wrong model can lead to spurious results. We have shown that ARGweaver-D is fairly robust to modest misspecifications in the model, but we recommend that careful demographic analysis be performed before running ARGweaver-D to ensure that the best possible model is used.
Finally, we detected small amounts of introgression for two additional events: Hum→Den and Sup→Nea. Because no previous evidence has been reported for these events, we expected that our predictions would be roughly in line with our false positive rates. Indeed, for the Hum→Den event, we predicted a slightly smaller fraction (0.37%) than the estimated false positive rate from simulations (0.41%). For Sup→Nea, however, we predicted 0.75% of the genome to be introgressed, which is slightly higher than the corresponding estimated false positive rate (0.65%). While these fractions are too small to draw strong conclusions, it is plausible that if Homo erectus mixed with the Denisovans, they may have also mixed with Neanderthals, perhaps in the Middle East; or perhaps DNA passed from Homo erectus to Neanderthal through the Denisovans. Altogether, given the number of gene flow events now documented among ancient hominins, it may be reasonable to assume that genetic exchange was likely whenever two groups overlapped in time and space.