|
|
Post by Admin on Nov 18, 2020 6:08:13 GMT
Human Coronavirus 229E (alphacoronavirus) Strain 229E was discovered during 1966, when researchers were characterizing five novel agents that were isolated from the respiratory tract of humans who had contracted the common cold [23]. 229E was adapted to grow in WI-38 lung fibroblast cell lines, and was later shown to be morphologically identical to IBV and MHV [24]. Symptoms of 229E infection include general malaise, headache, nasal discharge, sneezing, and a sore throat [25]. A small portion of patients (10–20%) will also exhibit fever and cough. The incubation time is approximately 2–5 days, followed by illness lasting between 2 and 18 days, and clinically indistinguishable from respiratory tract infections caused by other pathogens, such as rhinovirus and influenza A [18]. 229E is distributed globally (Figure 1A).  Human Coronavirus OC43 (betacoronavirus, lineage A) Strain OC43 was discovered, during 1967, in the nasopharyngeal wash of a patient with the common cold, and is adapted to grow in organ cultures containing suckling mouse brains. Similar to 229E, OC43 is morphologically indistinguishable from IBV and MHV [24], and patients infected with OC43 present the same clinical symptoms as that of 229E [24]. However, there is no serological cross-reactivity between 229E and OC43 [24]. OC43 is also distributed globally (Figure 1A). SARS-CoV (betacoronavirus, lineage B) Patients infected with SARS-CoV initially present with fever, myalgia, headache, malaise, and chills, followed by a nonproductive cough, dyspnea, and respiratory distress generally 5 to 7 days later, which may result in death (http://www.who.int/csr/sars/en/WHOconsensus.pdf?ua=1). Other notable features in some cases include infection of the gastrointestinal tract, liver, kidney, and brain. Diffuse alveolar damage, epithelial cell proliferation, and an increase in macrophages is seen in SARS-CoV infection of the lung. Lymphopenia, hemophagocytosis in the lung, in addition to white-pulp atrophy of the spleen observed in SARS patients, are similar to fatal H5N1 influenza virus infections [1]. Diarrhea is observed in approximately 30–40% of SARS infections (http://www.cdc.gov/sars/about/faq.html). An outbreak of disease caused by SARS-CoV, originating from Guangdong Province in southern China during November 2002, eventually spread to other countries in Asia, in addition to North America and Europe (37 countries/regions in total) over 9 months (http://www.who.int/ith/diseases/sars/en/) (Figure 1B). An eventual 8273 cases were reported, with 775 deaths for a case fatality rate (CFR) of 9%, and the majority of cases and deaths occurred in mainland China and in Hong Kong [19]. The elderly were more susceptible to SARS disease, with a mortality rate of over 50% (http://www.cdc.gov/sars/surveillance/absence.html). Human Coronavirus NL63 (alphacoronavirus) NL63 is primarily associated with young children, the elderly, and immunocompromised patients with respiratory illnesses [26]. NL63 was a novel HCoV isolated from a 7-month-old child with coryza, conjunctivitis, fever, and bronchiolitis in the Netherlands during late 2004 [26], and an independent investigation described the isolation of virtually the same virus from a nasal sample collected from an 8-month-old boy suffering from pneumonia in the Netherlands [27]. NL63 was also detected in New Haven, USA, during 2005 amongst 79 of 895 children, and was initially called HCoV-NH (NH), but genetic and phylogenetic analyses showed that NH and HKU1 likely are the same species [26, 28]. Infections with NL63 typically result in mild respiratory disease similar to the common cold, characterized by cough, rhinorrhea, tachypnea, fever, and hypoxia, and resolve on their own [29]. Obstructive laryngitis, also known as croup, is frequently observed with NL63 infections. A study estimated that NL63 accounts for an estimated 4.7% of common respiratory diseases [26]. NL63 is distributed globally (Figure 1A). Human Coronavirus HKU1 (betacoronavirus, lineage A) HKU1 was first discovered during January 2005 in a 71-year-old patient from Hong Kong who had been hospitalized with pneumonia and bronchiolitis [30]. The symptoms of HKU1 respiratory tract infections are not able to be separated from those caused by other respiratory viruses. Most patients present with fever, running nose, and cough for infections in the upper respiratory tract, whereas a fever, productive cough, and dyspnea are the common symptoms presenting for infections in the lower respiratory tract [31]. Most HKU1 infections are self-limiting, with only two deaths reported in patients with pneumonia due to HKU1 [32]. Although HKU1 is clinically relatively mild in children, HKU1 infection is associated with a high incidence of seizures, and was found in a patient with meningitis [33, 34]. HKU1 cases outside Asia were detected in New Haven, USA, in 2 out of 851 children [35], and also in Australia [36], France [37], and Brazil [38], indicating a global distribution for the virus (Figure 1A). MERS-CoV (betacoronavirus, lineage C) MERS-CoV was first isolated from the lungs of a 60-year-old patient who had died from a severe respiratory illness in Jeddah, Saudi Arabia, 2012 [39]. Clinical manifestations of MERS-CoV infection range from asymptomatic to severe pneumonia with acute respiratory distress, septic shock, and renal failure resulting in death [40]. A typical disease course begins with fever, cough, chills, sore throat, myalgia, and arthralgia, followed by dyspnea and rapid progression to pneumonia [40, 41, 42, 43]. Approximately one-third of patients present with gastrointestinal symptoms, such as diarrhea and vomiting. Acute renal impairment was the most striking feature of disease caused by MERS-CoV, which is thus far unique for human CoV infections [40, 44]. Seventy-five percent of patients with MERS disease also had at least one other comorbidity, and patients who died were more likely to have a pre-existing/underlying condition [40]. Countries around the Arabian Peninsula are known to be endemic for MERS-CoV, and Saudi Arabia has reported the most cases, but since its discovery in 2012, cases have been occasionally exported to other countries through travel, sometimes causing clusters of secondary outbreaks (Figure 1B) [45]. As of December 31, 2015, a total of 1621 laboratory-confirmed infections have been reported, with 584 deaths (CFR = 36.0%) over 26 countries (http://www.who.int/csr/disease/coronavirus_infections/en), making MERS-CoV one of the most dangerous viruses known to humans. Ecology of Human |
|
|
|
Post by Admin on Nov 18, 2020 21:22:09 GMT
Ecology of Human Coronaviruses Based on currently available evidence, 229E, OC43, NL63, and HKU1 are well adapted to humans, and the viruses widely circulate in the human population (Figure 2), with most cases causing mild disease in immunocompetent adults, and none of these viruses have been found to be maintained within an animal reservoir. However, in the case of SARS-CoV and MERS-CoV, the viruses are not as well adapted to being maintained in humans, and thus are likely spread mainly in zoonotic reservoir(s), with occasional spillover into the susceptible human population, possibly via an intermediate host species.  Figure 2 Intra- and Inter-Species Transmission of Human Coronaviruses. Red, yellow, green, blue, brown, and purple arrows represent transmission of MERS-CoV, SARS-CoV, NL63, HKU1, OC43, and 229E, respectively, between bats, camels, cows, humans, and masked palm civets (shown in a legend on the side of the figure). Unbroken arrows represent confirmed transmission between the two species in question, and broken arrows represent suspected transmission. Regarding SARS-CoV, epidemiology data implicated masked palm civets (Paguma larvata) from live animal markets (LAM) in Guangdong Province, China, as a route of exposure to SARS-CoV [46]. However, masked palm civets from the wild or from farms without LAM exposure were largely negative for SARS-CoV [47]. This suggests that palm civets were an intermediate host, but not a reservoir for SARS-CoV [48]. Subsequent studies have shown that wild horseshoe bats (Rhinolophidae family), which can also be found in LAM in China and served in some Chinese restaurants in Guangdong, China, have detectable levels of antibodies against SARS-CoV and a SARS-CoV-like virus (SARSr-Rh-Bat CoV) [49, 50], suggesting a bat origin for SARS-CoV. An evolutionary relationship between coronaviruses and bats was proposed, in which the ancestor for SARS-CoV first spread to bats of the Hipposideridae family, then Rhinolophidae, then masked palm civets and eventually humans [51]. Recently, two new SARS-like CoVs were isolated in horseshoe bats, and these viruses showed the highest relationship to SARS-CoV from all known bat coronaviruses [52]. ORF8 analysis of the SARS-like CoVs in bats suggests that Chinese horseshoe bats are the natural reservoirs of SARS-CoV [52, 53], and that intermediate hosts might not be needed for direct human infection, particularly for some bat SARS-CoV-like viruses [52]. In the case of MERS-CoV, studies in Oman, Saudi Arabia, Qatar, United Arab Emirates, and Jordan have shown that dromedary camels are seropositive for neutralizing antibodies against MERS-CoV [54], as well as camels of Middle East origin in Africa, including Egypt, Kenya, Nigeria, Ethiopia, Tunisia, Somalia, and Sudan [55]. Subsequent studies on dromedary camels in Saudi Arabia [56], Qatar [57], and Egypt [58] show that live MERS-CoV can be isolated primarily from the nasal swabs of camels, showing that camels are potential source of MERS-CoV infection. However, many confirmed cases lack contact history with camels [59], suggesting direct human-to-human transmission, or through contact with a yet-to-be-identified animal species that is maintaining MERS-CoV. Studies on HKU4, a coronavirus of bat origin and the most phylogenically closely related to MERS-CoV, had shown that HKU4 is able to utilize the CD26 receptor for virus entry [60]. As CD26 is a known receptor for MERS-CoV [61], the similarity in receptor specificity of these two CoVs supports the hypothesis that MERS-CoVs is also a bat-originated CoV. However, live MERS-CoV has not been isolated from wild bats, nor have viral sequences been detected. A recent investigation discovered that multiple HCoV species, including MERS-CoV, beta-CoV group A, and a 229E-like virus, circulate amongst dromedary camels in Saudi Arabia [62]. Furthermore, multiple lineages of circulating MERS-CoV in these camels had resulted in a dominant, recombinant MERS-CoV strain that was responsible for the MERS-CoV outbreaks during 2015 [62]. These results show that various CoVs of human and animal origin are currently circulating in the wild, providing ample opportunity for CoVs to undergo evolution and genetic recombination, thereby resulting in recombinant CoVs that may potentially be more deadly to humans. In the following sections, we summarize the evolution and genetic recombination of the current HCoVs. |
|
|
|
Post by Admin on Nov 19, 2020 6:00:22 GMT
Phylogeny, Evolution and Genetic Recombination in Human Coronaviruses Phylogenetic Analysis The phylogenetic tree for the CoVs of zoonotic and human origin was constructed based on the full genome. It can be observed that 229E and NL63 belong to the alpha-CoV genus and are grouped together with porcine epidemic diarrhea virus (PEDV), TGEV, porcine respiratory coronavirus (PRCV), feline infectious peritonitis virus (FIPV), and some bat-derived coronaviruses (Figure 3). The topology of the phylogeny is similar to the tree constructed based on the RNA-dependent RNA polymerase gene [55].  Figure 3 Phylogenetic Tree of Known Animal and Human Coronaviruses. Phylogenetic analysis of the full-length genome sequences of coronaviruses publicly available in GenBank was performed using RAxML, with 1000 bootstrap replicates. Lines with different colors represent different coronavirus species, Alpha-coronaviruses (blue lines), Beta-coronaviruses (red lines for Beta-coronavirus C, and black lines for Beta-coronavirus A, B, and D), Gamma-coronaviruses (green lines), and Delta-coronaviruses (purple lines). Human isolates are highlighted with different colors, whereas strains from other hosts are shown in black. Numbers at the branches represent bootstrap values obtained in the phylogenetic analysis. OC43, HKU1, SARS-CoV, and MERS-CoV belong to the beta-CoV genus. Lineage A includes OC43 and HKU1, MHV, porcine hemagglutinating encephalomyelitis virus (PHEV), in addition to equine, rabbit, camel, bovine, antelope-derived animal coronaviruses. Lineage B includes SARS-CoV, SARS-like viruses of bat and palm civet origin, and some bat-derived CoVs. Lineage C includes MERS-CoV and some bat-derived viruses. Lineage D contains only bat-derived coronavirus (Figure 3). In contrast to alpha- and beta-CoVs, gamma-CoVs consist mainly of avian coronaviruses (such as IBV), as well as CoVs isolated from aquatic animals, whales, and dolphins. CoVs of wild-bird origin are clustered into the delta-CoV, which also contains some swine-derived CoVs (Figure 3). In general, alpha- and beta-CoVs primarily include CoVs from mammals, whereas gamma- and delta-CoVs mostly include CoVs of avian origin. Notably, all currently known HCoVs belong to either the alpha- or betacoronavirus genera, which also contain CoVs of mainly bat origin. Mutation and Genetic Recombination in Coronaviruses The estimated mutation rates in CoV are moderate to high compared to other single-stranded RNA (ssRNA) viruses, and the average substitution rate for CoVs was ∼10−4 substitutions per year per site [63]. The nucleotide mutation rate of IBV for the hypervariable region in the S gene was estimated to be 0.3–0.6 × 10–2 per site per year [64]. The S gene of OC43 possessed an average rate of 6.410.58 × 10–4 substitution rates per site per year [65]. Additionally, the S gene of 229E possessed a rate of ∼3 × 10− 4 substitutions per site per year [63]. For SARS-CoV, the mutation rate in the whole genome was estimated to be 0.80–2.38 × 1–3 nucleotide substitutions per site per year, and the nonsynonymous and synonymous substitution rates were estimated to be 1.16–3.30 × 10–3 and 1.67–4.67 × 10–3 per site per year, respectively, which is similar to other RNA viruses [66]. The evolutionary rate for MERS-CoV genomes was estimated as 1.12 × 10−3 substitutions per site per year (95% credible interval [95% CI], 8.76 × 10−4; 1.37 × 10−3) [67]. Furthermore, the large RNA genome in CoV allows for extra plasticity in genome modification by mutations and recombinations, thereby increasing the probability for intraspecies variability, interspecies ‘host jump’, and novel CoVs to emerge under the right conditions [68, 69, 70]. The major reason for recombination may be at the replication step in the virus life cycle. During replication, a set of subgenomic RNAs is generated, increasing the homologous recombination rate among closely related genes from different lineages of CoVs or other viruses by template switching [33]. Concurrently, circulating CoVs in multiple host species likely contribute to increases in the rate of recombination events. However, the exact mechanism of genetic recombination in CoVs remains unclear: recombination site ‘breakpoints’ in the viral genome – the crossing point of the recombinant genes between two distinct viral strains or genotypes – appear to be random, as different recombinant strains have different breakpoints. Genetic recombination has been previously documented for animal CoVs, for instance: MHV [71], TGEV [72], as well as feline and canine coronaviruses [73, 74]. Genetic recombination for other HCoVs including OC43, NL63, HKU1, SARS-CoV [51], and MERS-CoV have also been documented and are discussed in detail below. OC43 has diverged into five distinct genotypes (A to E). Genotype A is typified by the prototype VR759 strain that was isolated in 1967 [75]. Genotypes B and C are two naturally circulating viruses. Genotype D emerged via recombination between genotypes B and C viruses at the breakpoint of 2500–5000 bp in the NSP2–NSP3 gene and 15500 bp-3′end in NSP12-N genes [75] (Figure 4, Key Figure). Genotype E, identified in 2014, emerged by recombination amongst the genotype B, C, and D viruses with different breakpoints in the genes compared to the generation of genotype D [76] (Figure 4). A study in France, between 2001 and 2013, demonstrated that OC43 strains have more recombination patterns in Lower Normandy, and circulating OC43 viruses have a high genetic diversity [17].  Figure 4 Key Figure: Genetic Recombination in Human Coronaviruses NL63 also has the potential for genetic recombination, and one such example is between the Amsterdam 1 and 496 strains. Recombination site breakpoints were identified in two parts of the S gene [63] (Figure 4). Recombination events have been observed for different genotypes of HKU1, in which recombination breakpoints were found on genotypes B and C in the nsp6-nsp7 and nsp16-HE genes, respectively [70] (Figure 4). HKU1 was also shown to be able to recombine with other animal beta-CoVs, such as MHV, at the nsp3, nsp4, nsp6, nsp7, nps9, and nsp10 genes [77] (Figure 4). SARS-CoV has a possible recombinant history with lineages of alpha- and gamma-CoV [78]. Many specific breakpoints and a large number of smaller recombinant regions had been identified in the RNA-dependent RNA polymerase (RDRP, nsp12 gene) at the following locations: nucleotides 13 392–13 610, 15 259–15 342, and 15 974–16 108 based on the sequence of the SARS-CoV TOR2 strain [78]. Recombination locations were also identified in the nsp9 and most of nsp10 gene located at 12 613–13 344, and parts of nsp14 (18 117–18 980) [78]. Another study analyzing the genome of the SARS-CoV CV7 strain had shown that seven putative recombination locations were found between SARS-CoV and six other coronaviruses: PEDV, TGEV, BCV, 229E, MHV, and IBV (Figure 4). This further suggests that SARS-CoV emerged via serial horizontal transmission and by genetic recombination, enhancing the adaptation process to its new host [79]. Recombination was also detected between the SARS-related bat-associated CoV (SARSr-Bat CoVs), in which recombination between the Rp3 strain from Guangxi Province and Rf1 strain from Hubei Province, China, generated a new strain (SARSr-Civet CoV SZ3) in civets, with a breakpoint at the nsp16/spike and S2 region [51] (Figure 4). Furthermore, the SARS-CoV Rf1/2004 strain is itself a recombinant from the bat CoV 279/2004, bat CoV Rm1/2004 and civet CoV SZ3/2003 [51] (Figure 4). A recent study showed that the epidemic MERS-CoV experienced recombination events between the different lineages, which occurred in dromedary camels in Saudi Arabia [62]. Lineages 3 and 5 viruses were predominant during July and December 2014; however, lineage 5 became dominant within the camel population by 2015. Lineage 5, which is closely associated with the MERS-CoV causing the outbreak in South Korea in 2015, as well as recent human infections in Riyadh, Saudi Arabia, is a recombinant virus between lineages 3 and 4, or groups 3 and 5 of clade B (Figure 4) [62, 80]. Concluding Remarks The result from a high frequency of recombination events in CoVs is the generation of novel viruses with a high genetic diversity, with unpredictable changes in virulence during human infections. With multiple species of CoVs circulating in the wild amongst different animal species that may constantly interact with one another, it is likely not a matter of if, but when, the next recombinant CoV will emerge and cause another outbreak in the human population. As such, some crucial future areas of investigation include: (i) the prevalence of HCoVs already circulating within the animal population, (ii) the commonality of coronavirus recombination in animals, (iii) animals which may potentially serve as mixing vessels for the generation of novel recombinant CoVs, and (iv) a surveillance network to monitor and predict the potential emergence of a highly virulent, recombinant CoV from animals (see Outstanding Questions). Furthermore, lessons from the SARS-CoV and MERS-CoV outbreaks must be urgently learned in advance to effectively prepare for the next CoV outbreak. |
|
|
|
Post by Admin on Nov 19, 2020 20:37:22 GMT
There are four common cold coronaviruses that we all catch at some stage. We generate antibodies to them, but our immune memory of them fades over time, and we get re-infected. Their names are all too easily forgotten—OC43, HKU1, 229E, and NL63—but our immune systems may nevertheless remember them for a time. There have been hints that exposure to these common coronaviruses might offer some protection from COVID-19, mostly by looking at signs of immune memory in blood samples taken from before the pandemic. A study in the Journal of Clinical Investigation reports the first clinical evidence linking recent endemic coronavirus infections to less severe COVID-19 and even a reduced death rate in patients. “The COVID-19 disease is actually much less severe in those patients who had documented endemic coronavirus infections.” —Manish Sagar, Boston Medical Center The authors at Boston University School of Medicine found evidence for this by poring over the medical records of thousands of patients who had visited Boston Medical Center as inpatients or outpatients, most probably for respiratory illnesses, between 2015 and 2020. Each person had been assessed for infection using a PCR test that screens for bacteria and viruses, including the four endemic coronaviruses. In total, 15,928 patients had at least one such PCR test. Of them, 875 tested positive for an endemic coronavirus (this group was called eCoV+), while the remaining 15,053 people never had a documented coronavirus infection (termed eCoV-). Of the entire cohort, a total of 1,812 (11.4 percent) later returned for a SARS-CoV-2 test during the initial COVID-19 surge in Boston between March 12 and June 12. “Our study is the first to examine people with known endemic coronavirus infections, and compare them to people who, as far as we know, don’t have any recent documented coronavirus infections,” says Manish Sagar, the lead author of the study and a virologist at Boston Medical Center. The infection rate for SARS-CoV-2 was no different between those who had a recently recorded endemic coronavirus infection (eCoV+) and those who did not have a positive test (eCoV-). This led the authors to conclude that a recent infection with endemic coronaviruses did not keep SARS-CoV-2 at bay—both groups were just as likely to become infected with the pandemic virus. When the researchers peered closer at the data, they observed an important difference between the two groups. “The COVID-19 disease is actually much less severe in those patients who had documented endemic coronavirus infections,” says Sagar. The odds of intensive care unit (ICU) admission were significantly lower in eCoV+ than in eCoV- patients, and there was “a trend towards lower odds of mechanical ventilation,” the authors write in their report. The data also show that among hospitalized patients who had previous positive test results for endemic coronavirus, 4.8 percent of them died compared with 17.7 percent among those in the group without such a test result. Local immune memory may help explain these results. Such “heterotypic immunity,” says immunologist Joseph Mizgerd, director of the pulmonary center at Boston University School of Medicine, occurs when immune memory is etched into the lungs and/or nose. It’s common after other types of respiratory infections and might offer protection against SARS-CoV-2 if elicited by endemic coronaviruses. Although the Boston group did not measure this type of immunity in patients, they now hypothesize that local immunity gained from endemic coronaviruses helps limit lung injury during COVID-19. “We are testing that in ongoing experiments,” Mizgerd says by email. He adds that such cross-reactive immunity is often mediated by memory T cells, which can localize in the lung, and he notes that lung-localized heterotypic T cells can prevent severe lung infection during pneumonias caused by other types of respiratory pathogens. If indeed prior infection does ramp up protection against SARS-CoV-2, the study could not answer how long it takes for any such benefit to taper off. Nor did the work shed light on which of the four endemic coronaviruses in particular might be offering protection against the pandemic virus. The scientists are seeking funding to expand their research and include data from other institutions. Mizgerd and his team did not look into which immune components may be responsible for an endemic coronavirus influencing a person’s immune response to SARS-CoV-2. This is something that immunologist Dennis Burton at the Scripps Research Institute in La Jolla, California, and his colleagues have investigated. Since the start of the pandemic, they have been interested in whether pre-existing immune responses to seasonal coronaviruses could influence antibody responses to SARS-CoV-2. In a study published in September as a preprint on bioRxiv, Burton and colleagues compared serum antibodies and antibody-producing B cells from 36 donors sampled prior to the pandemic to see whether those antibodies reacted with the spike protein from the new pandemic virus. Very few antibodies from before the pandemic reacted to SARS-CoV-2, the team found. The vast majority did not bind strongly to the new virus, although they did identify one antibody that could neutralize SARS-CoV-2. The group also detected memory B cells in blood samples from before the pandemic that were turned on by the presence of SARS-CoV-2. This activation triggered them to make antibodies that reacted against some proteins made by SARS-CoV-2. “That would suggest that there is some cross reactivity there,” says Burton. “A cross-protective vaccine that protects against SARS-CoV-2 plus the endemic coronaviruses would be a really great boon.” —Manish Sagar, Boston Medical Center A recent Science study reported that 5 percent of 302 adults and 43 percent of 48 children had antibodies that reacted against certain proteins produced by SARS-CoV-2. Children are more prone to common cold coronavirus infections, perhaps explaining why they might harbor such antibodies, and why they suffer less severe COVID-19 symptoms. “We do not know yet if the presence of such antibodies modifies the risk of becoming infected or the severity of disease,” senior author George Kassiotis at the Francis Crick Institute in London explains by email. There are conserved parts of the S2 peptide of the spike protein, such as the fusion peptide, in most coronaviruses “that are targeted by such cross-reactive and potentially cross-protective antibodies,” Kassiotis notes. This “may hold promise for a universal vaccine protecting against current, as well as future CoVs,” the authors write in their Science paper. Kassiotis says that concerns that “antibody immunity might be short-lived have now been allayed” by recent studies and adds that even if antibodies fell below detectable levels, “the cells that made them will still be there and will respond faster and better to re-infection.” Antibodies and B cells are part of only one aspect of our immune memory to viruses. Multiple investigations since the beginning of the pandemic have suggested that between 20 percent and 50 percent of people who had never encountered SARS-CoV-2 had T cells that nevertheless seemed to react against peptides from this virus, as noted recently in a Science paper. In another study in Nature, researchers in Singapore identified memory T cells in patients who had recovered from SARS back in 2003. These were reactive to proteins from SARS-CoV-2, supporting the idea that T cell memory from infections with human coronaviruses may play a role in the response to an infection with the new pandemic virus. An additional study recently published in Science used human blood samples from before the pandemic to locate parts of SARS-CoV-2 that stimulated existing T cells. The study found a range of memory T cells that could react to both the new virus and to the four common cold coronaviruses, again suggesting that existing T cells against common coronaviruses could play a role in the immune response to SARS-CoV-2 in some patients. Immunologist Stanley Perlman of the University of Iowa who was not involved in any of these studies says that “everybody should have memory B cells against common cold coronaviruses.” We may also have memory T cells that remember these viruses and perhaps help with fighting SAR-CoV-2. However, Perlman emphasized that the implication of this for COVID-19 “is still a work in progress.” Burton says he hopes to dig into a molecular understanding of the cross-reactivity of antibodies, which might help design a vaccine against not just SARS-CoV-2, but common cold coronaviruses too. These viruses usually cause mild symptoms, but not always. “A cross-protective vaccine that protects against SARS-CoV-2 plus the endemic coronaviruses would be a really great boon,” says Sagar. “These coronaviruses are causes of the common cold, but they are also really important causes of pneumonia, pneumonia hospitalizations, and pneumonia deaths.” |
|
|
|
Post by Admin on Nov 20, 2020 4:24:08 GMT
Recent endemic coronavirus infection is associated with less severe COVID-19 Manish Sagar,1 Katherine Reifler,1 Michael Rossi,1 Nancy S. Miller,2 Pranay Sinha,1 Laura White,3 and Joseph P. Mizgerd1 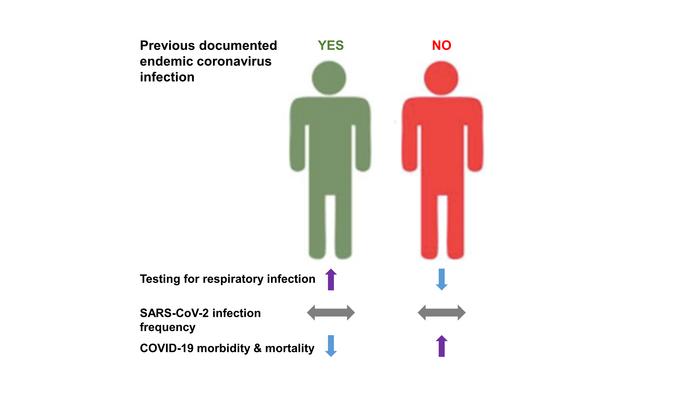 Abstract Four different endemic coronaviruses (eCoVs) are etiologic agents for the seasonal “common cold,” and these eCoVs share extensive sequence homology with human severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). Here, we show that individuals with as compared to without a relatively recent documented eCoV were tested at greater frequency for respiratory infections but had similar rate of SARS-CoV-2 acquisition. Importantly, the patients with a previously detected eCoV had less severe coronavirus disease-2019 (COVID-19) illness. Our observations suggest that pre-existing immune responses against endemic human coronaviruses can mitigate disease manifestations from SARS-CoV-2 infection. Introduction While severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) emerged recently, other coronaviruses are endemic in the human population. Four different human coronaviruses (HCoV-OC43, HCoVHKU1, HCoV-NL63, and HCoV-229E) are among the most common etiologic agents for the seasonal “common cold” and also cause pneumonia (1, 2). SARS-CoV-2 induced disease, termed coronavirus disease 2019 (COVID-19), can vary from asymptomatic to acute respiratory distress syndrome requiring mechanical ventilation and often leading to death (3, 4). The endemic coronaviruses (eCoVs) share extensive sequence homology with SARS-CoV-2, and immune responses to the eCoVs can cross-react with SARS-CoV-2 antigens (5-8). Whether prior infections with eCoV elicits immunologic memory that influences SARS-CoV-2 acquisition and COVID-19 outcomes remains uncertain. |
|

