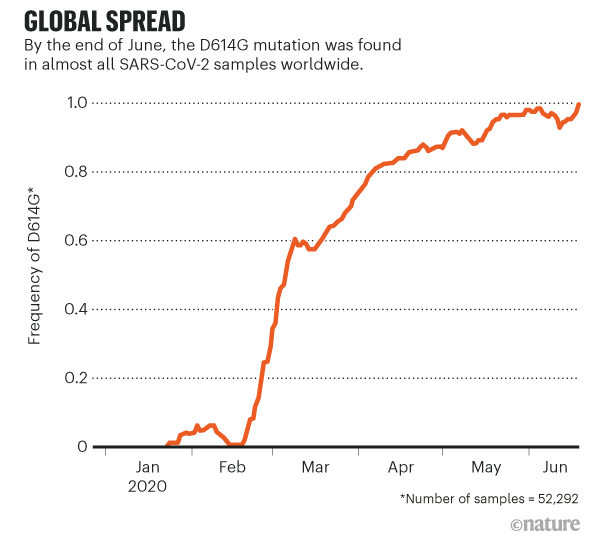
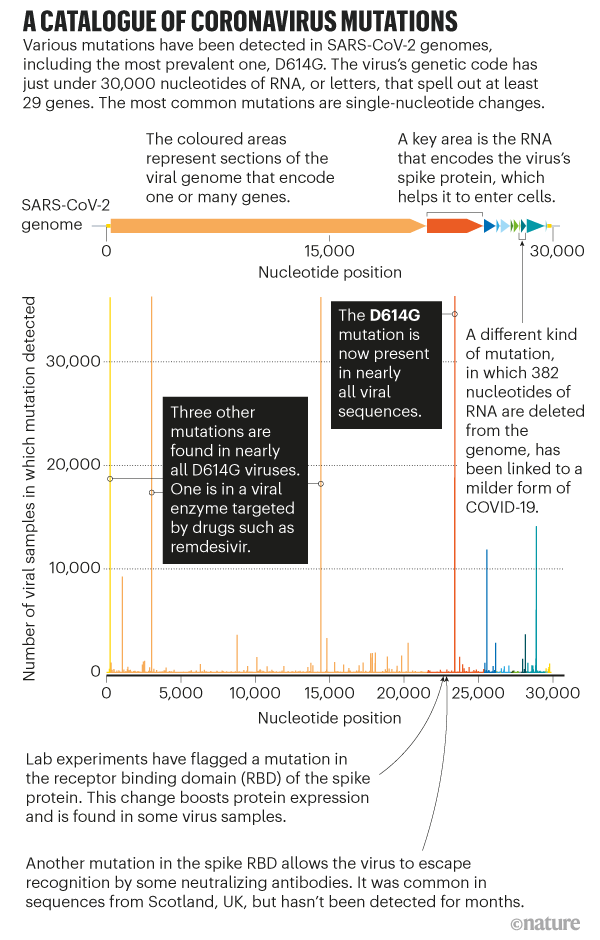
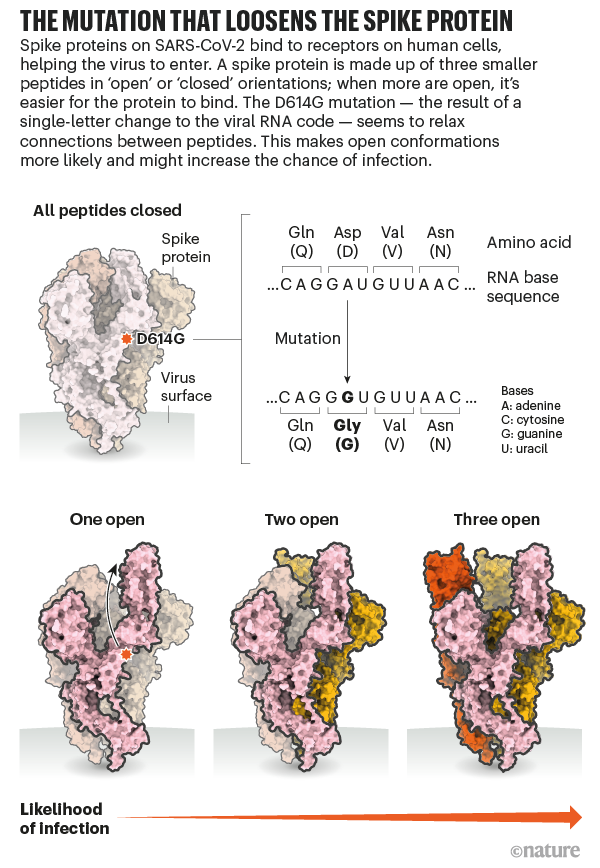
Post by Admin on Aug 28, 2020 7:59:02 GMT
Structural Implications of the Spike D614G Change
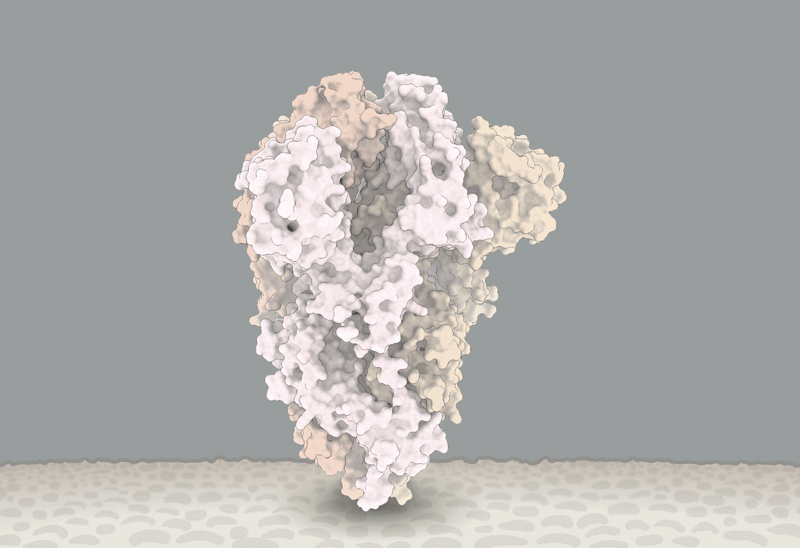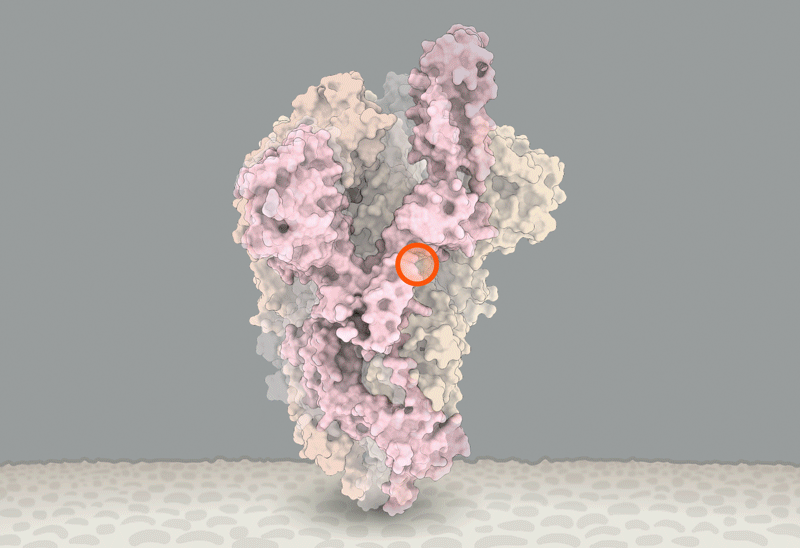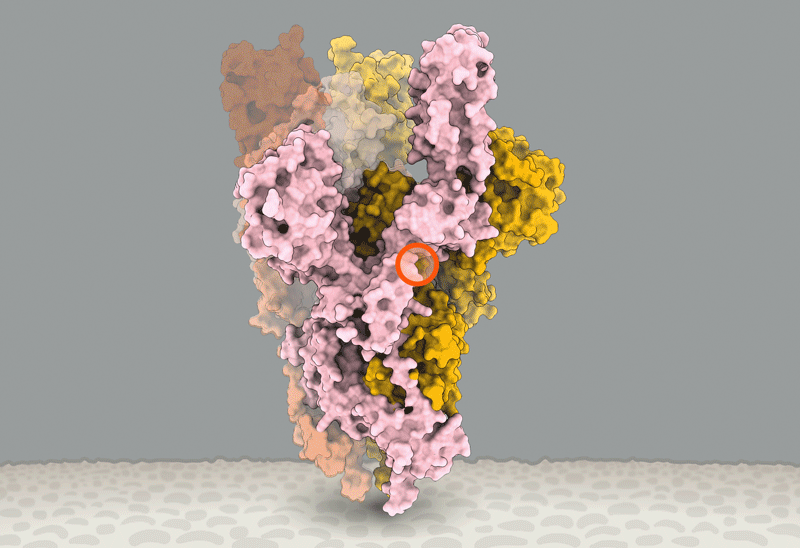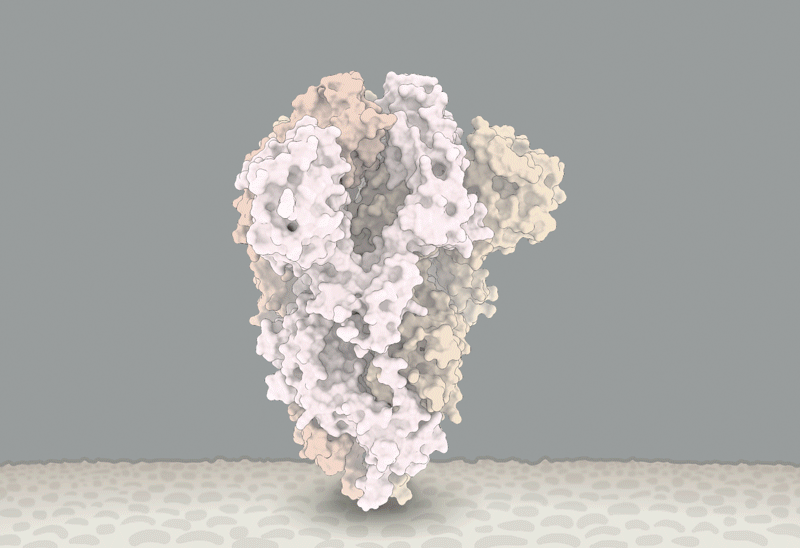
D614 is located on the surface of the Spike protein protomer, where it can form contacts with the neighboring protomer (Figure 4A). Cryoelectron microscopy (cryo-EM) structures (Walls et al., 2020; Wrapp et al., 2020) indicate that the side chains of D614 and T859 of the neighboring protomer (Figure 4B) form a between-protomer hydrogen bond, bringing together a residue from the S1 unit of one protomer and a residue of the S2 unit of the other protomer (Figure 4C). The change to G614 would eliminate this side-chain hydrogen bond, possibly increasing main-chain flexibility and altering between-protomer interactions. In addition, this substitution could modulate glycosylation at the nearby N616 site, influence the dynamics of the spatially proximal fusion peptide (Figure 4D) of the neighboring protomer, or have other effects.

Figure 4. Structural Mapping of Amino Acid Changes and Clusters of Variation in the Spike Protein
(A) Sites including Spike 614 and those noted in Figure 7 mapped onto S1 and S2 units of the Spike protein (PDB: 6VSB). S1 and S2 are defined based on the furin cleavage site (protomer 1: S1, dark blue; S2, light blue; protomer 2: S1, dark green; S2, light green; protomer 3: gray). The RBD of protomer 1 is in the “up” position for engagement with the ACE2 receptor. Sites of interest are indicated by red balls, and variable clusters are labeled in red. The missing RBD residues at the ACE2 interface are shown in (D).
(B) Proximity of D614 (red) to an N-linked glycosylation sequon of its own protomer (blue) and to residues T859 and Q853 of the neighboring protomer (green) are shown. Black dashed lines indicate possible hydrogen bond formation. Dotted lines indicate the structurally unresolved region of the fusion peptide connecting to Q853.
(C) Schematic representation of potential protomer-protomer interactions shown in (B), in which D614 (red) from the S1 unit of one protomer (blue) is brought close to T859 from the S2 unit of the neighboring protomer.
(D) Sites of interest (red, residues 475–483) near the RBD (blue)-ACE2 (yellow) binding interface. The interfacial region is shown as a molecular surface (PDB: 6M17).
(E) Variable cluster 936–940 (red) in the HR1 region of S2. These residues occur in a region that undergoes conformational transition during fusion; pre-fusion (PDB: 6VSB) and post-fusion (PDB: 6LXT) conformations of HR1 are shown (left and right).
G614 Is Associated with Potentially Higher Viral Loads in COVID-19 Patients but Not with Disease Severity
SARS-CoV-2 sequences from 999 individuals presenting with COVID-19 disease at the Sheffield Teaching Hospitals NHS Foundation Trust were available and linked to clinical data. The Sheffield data include age, sex, date of sampling, hospitalization status (defined as outpatient [OP], inpatient [IP], requiring hospitalization, or admittance to the intensive care unit [ICU]), and the cycle threshold (Ct) for a positive signal in E-gene based RT-PCR. The Ct is used here as a surrogate for relative viral loads; lower Ct values indicate higher viral loads (Corman et al., 2020), but not all viral nucleic acids represent infectious viral particles. RT-PCR methods changed during the course of the study because of limited availability of testing kits. The first method involved nucleic acid extraction and the second method heat treatment (Fomsgaard and Rosenstierne, 2020). A generalized linear model (GLM) used to predict the PCR Ct based on the RT-PCR method, sex, age, and D614G status showed only the RT-PCR method (p < 2e−16) and D614G status (p = 0.037) to be statistically significant (Figure 5A). Lower Ct values were observed in G614 infections. While our paper was in revision, G614-variant association with low Ct values in vivo (Figure 5) was reported independently by two other groups (Lorenzo-Redondo et al., 2020; Wagner et al., 2020) in preprints that have not yet been peer reviewed.

Figure 5. Clinical Status and D614G Associations Based on 999 Subjects with COVID-19 and Linked Sequence and Clinical Data Were Sampled in Sheffield, England
(A) G614 was associated with a lower cycle threshold (Ct) required for detection; lower values are indicative of higher viral loads. The PCR method was changed partway through April 2020 because of shortages of nucleic acid extraction kits (Fomsgaard and Rosenstierne, 2020). The Ct levels for the two PCR methods (nucleic acid extraction versus simple heat inactivation) differ, and so we used a GLM to evaluate the statistical effect of D614G across methods.
(B) D614G status was not statistically associated with hospitalization status (outpatient [OP], inpatient [IP], or ICU) as a marker of disease severity, but age was highly correlated. The number of counts in each category is noted in the top right corner of each graph. See the main text and STAR Methods for statistical details.
We found no significant association between D614G status and disease severity as measured by hospitalization outcomes. A comparison of D614G status and hospitalization (combining IP and ICU) was not significant (p = 0.66, Fisher’s exact test), although comparing ICU admission with IP and OP did have borderline significance (p = 0.047) (Figure 5B). Regression analysis reinforced the result that G614 status was not associated with greater levels of hospitalization but that higher age (Dowd et al., 2020; Promislow, 2020), male sex (Conti and Younes, 2020; Promislow, 2020) and higher Ct values (lower viral loads) were highly predictive of hospitalization. Further analysis showed that viral load was not masking a potential D614G status effect on hospitalization (STAR Methods). Univariate analysis also found highly significant associations between age and male sex and hospitalization (STAR Methods).
G614 Is Associated with Higher Infectious Titers of Spike-Pseudotyped Virus
We quantified the infectious titers of pseudotyped single-cycle vesicular stomatitis virus (VSV) and lentiviral particles displaying D614 or G614 SARS-CoV2 Spike protein. For the VSV and lentiviral pseudotypes, G614-bearing viruses had significantly higher infectious titers (2.6- to 9.3-fold increase) than their D614 counterparts; this was confirmed in multiple cell types (Figures 6A–6C). Similar results, reported recently in a preprint that has not yet been peer reviewed, also suggest that G614 increases Spike stability and membrane incorporation (Zhang et al., 2020).

Figure 6. Viral Infectivity and D614G Associations
(A) A recombinant VSV pseudotyped with the G614 Spike grows to a higher titer than D614 Spike in Vero, 293T-ACE2, and 293T-ACE2-TMPRSS2 cells, as measured in terms of focus-forming units (ffu). ∗∗∗∗p < 0.0001 by Student’s t test in pairwise comparisons. Experiments were repeated twice, each time in triplicate. Using a GLM to assess viral infectivity of the D614 and G614 variants across cell types and to account for repeat experiments, we found that the G614 variant had an average 3-fold higher infectious titer than D614 and that this difference was highly significant (p = 9 × 10−11) (STAR Methods).
(B and C) Recombinant lentiviruses pseudotyped with the G614 Spike were more infectious than corresponding D614 S-pseudotyped viruses in (B) 293T/ACE2 (6.5-fold increase) and (C) TZM-bl/ACE2 cells (2.8-fold increase, p < 0.0001). Relative luminescence units (RLUs) of Luc reporter gene expression (Naldini et al., 1996) were standardized to the p24 content of the pseudoviruses (p24 content of pseudoviruses for 293T/ACE2 cells: D614 = 269 ng/mL, G614 = 255 ng/mL; p24 content of pseudoviruses for TZM-bl/ACE2 cells, D614 = 680 ng/mL, G614 = 605 ng/mL). Background RLUs were measured in wells that received cells but no pseudovirus.
(D and E) Convalescent serum from six individuals in San Diego (Donors A–F) can neutralize D614-bearing (orange) and G614-bearing (blue) VSV pseudoviruses. Percent relative infection is plotted versus log polyclonal antibody concentration. Error bars indicate the mean ± SD of two biological replicates, each having two technical replicates. In (D), the sensitivity to each form is shown seperately for each sera. In (E) the responses to all sera are combined in one graph, and two negative control normal human sera are indicated in grey.
TMPRSS2, a type-II transmembrane serine protease, cleaves the viral Spike after receptor binding to enhance entry of MERS-CoV, SARS-CoV, and SARS-CoV-2 (Hoffmann et al., 2020b; Kleine-Weber et al., 2018; Matsuyama et al., 2020; Millet and Whittaker, 2014; Park et al., 2016; Shulla et al., 2011; Zang et al., 2020). Spike 614 is in a pocket adjacent to the fusion peptide near the expected TMPRSS2 cleavage site, suggesting that there could be differences in the propensity and/or requirement for TMPRSS2 of the G614 variant. To test this hypothesis, we infected 293T cells stably expressing the ACE2 receptor in the presence or absence of TMPRSS2 and quantified the titer of infectious virus. We found similar fold changes in titers between D614 and G614 regardless of TMPRSS2 expression (Figure 6A). Hence, entry of G614-bearing viruses into 293T-ACE2 cells compared with D614-bearing viruses is not enhanced by TMPRSS2. Further studies are required to determine whether the G614 variant shows increased titers in lung cells, which may recapitulate native protease expression levels more faithfully, and to determine whether this variant increases the fitness of authentic SARS-CoV-2.
We also tested whether the D614G variations would be similarly neutralized by a polyclonal antibody. Convalescent sera of six San Diego residents, likely infected in early to mid-March 2020, when D614 and G614 were circulating, demonstrate equivalent or better neutralization of a G614-bearing pseudovirus compared with a D614-bearing pseudovirus (Figures 6D and 6E). Although we do not know with which virus each of these individuals were infected, these initial data suggest that, despite increased fitness in cell culture, G614-bearing virions are not intrinsically more resistant to neutralization by convalescent sera.
Additional Sites of Interest in the Spike Gene with Rare Mutations
Spike has very few mutations overall. A small set has reached 0.3% or more of the global population sample, the threshold for automatic tracking at the cov.lanl.gov website (Figures 7A and 7B; details are provided in Table S2). Regions in the alignment where entropy is relatively high compared with the rest of Spike (i.e., local clusters of rare mutations) are also tracked (Table S2). Genetic mutations of interest are mapped as amino acid changes onto a Spike structure (Figure 4). The mutation resulting in the signal peptide L5F change recurs many times in the tree and is stably maintained in about 0.6% of the global GISAID data. There are several clusters of mutations in the region of the Spike gene encoding the N-terminal domain (NTD) and RBD that are potential targets for neutralizing antibodies (Chen et al., 2017; Zhou et al., 2019; Sui et al., 2008; Tang et al., 2014; ter Meulen et al., 2006). The RBD cluster (positives 475–483) spans two positions, 475 and 476, that are located within 4 Å of bound ACE2 (Figure 4D; Yan et al., 2020). The fusion peptide contains a cluster of amino acid changes between 826–839; this cluster is highlighted in Figure 7 to illustrate our web-based tools for tracking variation (Figures 7A–7C). The fusion core of HR1 (Xia et al., 2020), next to the helix break in pre-fusion Spike, also contains a cluster of amino acid changes between 936-940 (Figure 4E). The motif SXSS (937–940) may enhance the association of helices (Dawson et al., 2002; Salamango and Johnson, 2015). The cytoplasmic tail of Spike also contains a site of interest, P1263L.

Figure 7. Tracking Variation in Spike
(A) Spike sites of interest (with a minimum frequency of 0.3% variant amino acids) are mapped onto a parsimony tree (for D614G; Figure S6). L5F recurs throughout the tree and is often clustered in small local clades. A829T is found in a single lineage. Other sites of interest cluster in a main lineage but are occasionally found in other parts of the tree in distant geographic regions and, thus, likely to recur at a low level. Build parsimony trees. A brief parsimony search (parsimony ratchet with 5 replicates) is performed with “oblong” (Goloboff, 2014). This is intended as an efficient clustering procedure rather than an explicit attempt to achieve accurate phylogenetic reconstruction, but it appears to yield reasonable results in this situation of a very large number of sequences with a very small number of changes, where more complex models may be subject to overfitting. When multiple most-parsimonious trees are found, only the shortest of these (under a p-distance criterion) is retained. Distance scoring is performed with PAUP∗ (Swofford, 2003).
(B) The global frequency of amino acid variants in sites of interest and the place where they are most commonly found. Such information could be useful if a vaccine or antibody is intended for use in a geographic region with a commonly circulating variant, so it could be experimentally evaluated prior to testing the planned intervention.
(C) Examples of exploratory plots showing A829T in Thailand and D839Y in New Zealand. Such plots for any variant in any region can be readily created at cov.lanl.gov/ to enable monitoring local frequency changes.
(D) Contiguous regions of relatively high entropy in the Spike alignment, indicative of local clusters of amino acid variation in the protein. The fusion-peptide cluster is used as example. It spans two sites of interest, labeled in blue and purple in (B) and (C). The alignment is created with AnalyzeAlign. Contemporary versions of these figures can be created at cov.lanl.gov/. Care should be taken to try to avoid systematic sequencing errors and processing artifacts among rare variants (for example, see Figure S7; De Maio et al., 2020; Freeman et al., 2020).
D614 is located on the surface of the Spike protein protomer, where it can form contacts with the neighboring protomer (Figure 4A). Cryoelectron microscopy (cryo-EM) structures (Walls et al., 2020; Wrapp et al., 2020) indicate that the side chains of D614 and T859 of the neighboring protomer (Figure 4B) form a between-protomer hydrogen bond, bringing together a residue from the S1 unit of one protomer and a residue of the S2 unit of the other protomer (Figure 4C). The change to G614 would eliminate this side-chain hydrogen bond, possibly increasing main-chain flexibility and altering between-protomer interactions. In addition, this substitution could modulate glycosylation at the nearby N616 site, influence the dynamics of the spatially proximal fusion peptide (Figure 4D) of the neighboring protomer, or have other effects.
Figure 4. Structural Mapping of Amino Acid Changes and Clusters of Variation in the Spike Protein
(A) Sites including Spike 614 and those noted in Figure 7 mapped onto S1 and S2 units of the Spike protein (PDB: 6VSB). S1 and S2 are defined based on the furin cleavage site (protomer 1: S1, dark blue; S2, light blue; protomer 2: S1, dark green; S2, light green; protomer 3: gray). The RBD of protomer 1 is in the “up” position for engagement with the ACE2 receptor. Sites of interest are indicated by red balls, and variable clusters are labeled in red. The missing RBD residues at the ACE2 interface are shown in (D).
(B) Proximity of D614 (red) to an N-linked glycosylation sequon of its own protomer (blue) and to residues T859 and Q853 of the neighboring protomer (green) are shown. Black dashed lines indicate possible hydrogen bond formation. Dotted lines indicate the structurally unresolved region of the fusion peptide connecting to Q853.
(C) Schematic representation of potential protomer-protomer interactions shown in (B), in which D614 (red) from the S1 unit of one protomer (blue) is brought close to T859 from the S2 unit of the neighboring protomer.
(D) Sites of interest (red, residues 475–483) near the RBD (blue)-ACE2 (yellow) binding interface. The interfacial region is shown as a molecular surface (PDB: 6M17).
(E) Variable cluster 936–940 (red) in the HR1 region of S2. These residues occur in a region that undergoes conformational transition during fusion; pre-fusion (PDB: 6VSB) and post-fusion (PDB: 6LXT) conformations of HR1 are shown (left and right).
G614 Is Associated with Potentially Higher Viral Loads in COVID-19 Patients but Not with Disease Severity
SARS-CoV-2 sequences from 999 individuals presenting with COVID-19 disease at the Sheffield Teaching Hospitals NHS Foundation Trust were available and linked to clinical data. The Sheffield data include age, sex, date of sampling, hospitalization status (defined as outpatient [OP], inpatient [IP], requiring hospitalization, or admittance to the intensive care unit [ICU]), and the cycle threshold (Ct) for a positive signal in E-gene based RT-PCR. The Ct is used here as a surrogate for relative viral loads; lower Ct values indicate higher viral loads (Corman et al., 2020), but not all viral nucleic acids represent infectious viral particles. RT-PCR methods changed during the course of the study because of limited availability of testing kits. The first method involved nucleic acid extraction and the second method heat treatment (Fomsgaard and Rosenstierne, 2020). A generalized linear model (GLM) used to predict the PCR Ct based on the RT-PCR method, sex, age, and D614G status showed only the RT-PCR method (p < 2e−16) and D614G status (p = 0.037) to be statistically significant (Figure 5A). Lower Ct values were observed in G614 infections. While our paper was in revision, G614-variant association with low Ct values in vivo (Figure 5) was reported independently by two other groups (Lorenzo-Redondo et al., 2020; Wagner et al., 2020) in preprints that have not yet been peer reviewed.
Figure 5. Clinical Status and D614G Associations Based on 999 Subjects with COVID-19 and Linked Sequence and Clinical Data Were Sampled in Sheffield, England
(A) G614 was associated with a lower cycle threshold (Ct) required for detection; lower values are indicative of higher viral loads. The PCR method was changed partway through April 2020 because of shortages of nucleic acid extraction kits (Fomsgaard and Rosenstierne, 2020). The Ct levels for the two PCR methods (nucleic acid extraction versus simple heat inactivation) differ, and so we used a GLM to evaluate the statistical effect of D614G across methods.
(B) D614G status was not statistically associated with hospitalization status (outpatient [OP], inpatient [IP], or ICU) as a marker of disease severity, but age was highly correlated. The number of counts in each category is noted in the top right corner of each graph. See the main text and STAR Methods for statistical details.
We found no significant association between D614G status and disease severity as measured by hospitalization outcomes. A comparison of D614G status and hospitalization (combining IP and ICU) was not significant (p = 0.66, Fisher’s exact test), although comparing ICU admission with IP and OP did have borderline significance (p = 0.047) (Figure 5B). Regression analysis reinforced the result that G614 status was not associated with greater levels of hospitalization but that higher age (Dowd et al., 2020; Promislow, 2020), male sex (Conti and Younes, 2020; Promislow, 2020) and higher Ct values (lower viral loads) were highly predictive of hospitalization. Further analysis showed that viral load was not masking a potential D614G status effect on hospitalization (STAR Methods). Univariate analysis also found highly significant associations between age and male sex and hospitalization (STAR Methods).
G614 Is Associated with Higher Infectious Titers of Spike-Pseudotyped Virus
We quantified the infectious titers of pseudotyped single-cycle vesicular stomatitis virus (VSV) and lentiviral particles displaying D614 or G614 SARS-CoV2 Spike protein. For the VSV and lentiviral pseudotypes, G614-bearing viruses had significantly higher infectious titers (2.6- to 9.3-fold increase) than their D614 counterparts; this was confirmed in multiple cell types (Figures 6A–6C). Similar results, reported recently in a preprint that has not yet been peer reviewed, also suggest that G614 increases Spike stability and membrane incorporation (Zhang et al., 2020).
Figure 6. Viral Infectivity and D614G Associations
(A) A recombinant VSV pseudotyped with the G614 Spike grows to a higher titer than D614 Spike in Vero, 293T-ACE2, and 293T-ACE2-TMPRSS2 cells, as measured in terms of focus-forming units (ffu). ∗∗∗∗p < 0.0001 by Student’s t test in pairwise comparisons. Experiments were repeated twice, each time in triplicate. Using a GLM to assess viral infectivity of the D614 and G614 variants across cell types and to account for repeat experiments, we found that the G614 variant had an average 3-fold higher infectious titer than D614 and that this difference was highly significant (p = 9 × 10−11) (STAR Methods).
(B and C) Recombinant lentiviruses pseudotyped with the G614 Spike were more infectious than corresponding D614 S-pseudotyped viruses in (B) 293T/ACE2 (6.5-fold increase) and (C) TZM-bl/ACE2 cells (2.8-fold increase, p < 0.0001). Relative luminescence units (RLUs) of Luc reporter gene expression (Naldini et al., 1996) were standardized to the p24 content of the pseudoviruses (p24 content of pseudoviruses for 293T/ACE2 cells: D614 = 269 ng/mL, G614 = 255 ng/mL; p24 content of pseudoviruses for TZM-bl/ACE2 cells, D614 = 680 ng/mL, G614 = 605 ng/mL). Background RLUs were measured in wells that received cells but no pseudovirus.
(D and E) Convalescent serum from six individuals in San Diego (Donors A–F) can neutralize D614-bearing (orange) and G614-bearing (blue) VSV pseudoviruses. Percent relative infection is plotted versus log polyclonal antibody concentration. Error bars indicate the mean ± SD of two biological replicates, each having two technical replicates. In (D), the sensitivity to each form is shown seperately for each sera. In (E) the responses to all sera are combined in one graph, and two negative control normal human sera are indicated in grey.
TMPRSS2, a type-II transmembrane serine protease, cleaves the viral Spike after receptor binding to enhance entry of MERS-CoV, SARS-CoV, and SARS-CoV-2 (Hoffmann et al., 2020b; Kleine-Weber et al., 2018; Matsuyama et al., 2020; Millet and Whittaker, 2014; Park et al., 2016; Shulla et al., 2011; Zang et al., 2020). Spike 614 is in a pocket adjacent to the fusion peptide near the expected TMPRSS2 cleavage site, suggesting that there could be differences in the propensity and/or requirement for TMPRSS2 of the G614 variant. To test this hypothesis, we infected 293T cells stably expressing the ACE2 receptor in the presence or absence of TMPRSS2 and quantified the titer of infectious virus. We found similar fold changes in titers between D614 and G614 regardless of TMPRSS2 expression (Figure 6A). Hence, entry of G614-bearing viruses into 293T-ACE2 cells compared with D614-bearing viruses is not enhanced by TMPRSS2. Further studies are required to determine whether the G614 variant shows increased titers in lung cells, which may recapitulate native protease expression levels more faithfully, and to determine whether this variant increases the fitness of authentic SARS-CoV-2.
We also tested whether the D614G variations would be similarly neutralized by a polyclonal antibody. Convalescent sera of six San Diego residents, likely infected in early to mid-March 2020, when D614 and G614 were circulating, demonstrate equivalent or better neutralization of a G614-bearing pseudovirus compared with a D614-bearing pseudovirus (Figures 6D and 6E). Although we do not know with which virus each of these individuals were infected, these initial data suggest that, despite increased fitness in cell culture, G614-bearing virions are not intrinsically more resistant to neutralization by convalescent sera.
Additional Sites of Interest in the Spike Gene with Rare Mutations
Spike has very few mutations overall. A small set has reached 0.3% or more of the global population sample, the threshold for automatic tracking at the cov.lanl.gov website (Figures 7A and 7B; details are provided in Table S2). Regions in the alignment where entropy is relatively high compared with the rest of Spike (i.e., local clusters of rare mutations) are also tracked (Table S2). Genetic mutations of interest are mapped as amino acid changes onto a Spike structure (Figure 4). The mutation resulting in the signal peptide L5F change recurs many times in the tree and is stably maintained in about 0.6% of the global GISAID data. There are several clusters of mutations in the region of the Spike gene encoding the N-terminal domain (NTD) and RBD that are potential targets for neutralizing antibodies (Chen et al., 2017; Zhou et al., 2019; Sui et al., 2008; Tang et al., 2014; ter Meulen et al., 2006). The RBD cluster (positives 475–483) spans two positions, 475 and 476, that are located within 4 Å of bound ACE2 (Figure 4D; Yan et al., 2020). The fusion peptide contains a cluster of amino acid changes between 826–839; this cluster is highlighted in Figure 7 to illustrate our web-based tools for tracking variation (Figures 7A–7C). The fusion core of HR1 (Xia et al., 2020), next to the helix break in pre-fusion Spike, also contains a cluster of amino acid changes between 936-940 (Figure 4E). The motif SXSS (937–940) may enhance the association of helices (Dawson et al., 2002; Salamango and Johnson, 2015). The cytoplasmic tail of Spike also contains a site of interest, P1263L.
Figure 7. Tracking Variation in Spike
(A) Spike sites of interest (with a minimum frequency of 0.3% variant amino acids) are mapped onto a parsimony tree (for D614G; Figure S6). L5F recurs throughout the tree and is often clustered in small local clades. A829T is found in a single lineage. Other sites of interest cluster in a main lineage but are occasionally found in other parts of the tree in distant geographic regions and, thus, likely to recur at a low level. Build parsimony trees. A brief parsimony search (parsimony ratchet with 5 replicates) is performed with “oblong” (Goloboff, 2014). This is intended as an efficient clustering procedure rather than an explicit attempt to achieve accurate phylogenetic reconstruction, but it appears to yield reasonable results in this situation of a very large number of sequences with a very small number of changes, where more complex models may be subject to overfitting. When multiple most-parsimonious trees are found, only the shortest of these (under a p-distance criterion) is retained. Distance scoring is performed with PAUP∗ (Swofford, 2003).
(B) The global frequency of amino acid variants in sites of interest and the place where they are most commonly found. Such information could be useful if a vaccine or antibody is intended for use in a geographic region with a commonly circulating variant, so it could be experimentally evaluated prior to testing the planned intervention.
(C) Examples of exploratory plots showing A829T in Thailand and D839Y in New Zealand. Such plots for any variant in any region can be readily created at cov.lanl.gov/ to enable monitoring local frequency changes.
(D) Contiguous regions of relatively high entropy in the Spike alignment, indicative of local clusters of amino acid variation in the protein. The fusion-peptide cluster is used as example. It spans two sites of interest, labeled in blue and purple in (B) and (C). The alignment is created with AnalyzeAlign. Contemporary versions of these figures can be created at cov.lanl.gov/. Care should be taken to try to avoid systematic sequencing errors and processing artifacts among rare variants (for example, see Figure S7; De Maio et al., 2020; Freeman et al., 2020).