|
|
Post by Admin on May 11, 2020 5:47:00 GMT
 From an evolutionary standpoint, it was not long ago that the Tsimshian people of modern-day Alaska and British Columbia were first confronted with European settlers—roughly 175 years, a mere handful of generations out of the Tsimshian’s 6,000-year American history. But that fateful encounter, which introduced smallpox and other alien ailments into their population, decimated the Tsimshian and threatened to compromise their genetic diversity in the years ahead. This landmark moment in Native American history captured the imagination of John Lindo, a genetic anthropologist at Emory University who delved deep into Tsimshian DNA as lead author on a just-published paper in the American Journal of Human Genetics. Lindo focused his research on the Tsimshian in an effort to understand the genetic dynamics surrounding their population collapse, which could shed light on the experience of many other Native American groups upon first contact with Europeans. :focal(319x195:320x196)/https://public-media.si-cdn.com/filer/bb/4e/bb4eb06e-9e05-48ad-b0ee-dbb2cdcddac2/tsimshian1.jpg) Employing cutting-edge genomic analysis, Lindo and his team compared modern Tsimshian DNA (obtained with consent from Tsimshian residents of Prince Rupert Harbour, Canada) against DNA found in millennia-old ancestral specimens (exhumed under community supervision and housed in the Canadian Museum of History), correcting for the degradation of the ancient DNA over time. What the researchers learned about the Tsimshian—on both sides of the fateful 19th-century population collapse—adds considerable nuance to the genetic and social history of a prominent First Nations people. What most surprised researchers was that the population of the ancient Tsimshian people was in decline long before the arrival of Europeans. Slowly and steadily, since their first settlement in modern Canada, the Tsimshian had been decreasing in number, not expanding as one might presume.  “We were completely expecting to see the population expand after that founding effect, when they entered from the Bering Strait,” Lindo says. “It was a big surprise to see that the population was on a steady decline before European contact.” For Lindo, this finding drives home a valuable lesson: all Native American peoples have their own stories to tell, and academics do a disservice when they proffer sweeping assertions. “Native Americans all have unique evolutionary histories,” he says. “They can’t just be summed up as ‘one race’ of Native Americans all experiencing the same thing after entering the Americas.” Many Native American populations swelled following their establishment, but the Tsimshian evidently took a different course. The eventual arrival of disease-bearing Europeans in the region ratcheted up the Tsimshian decline to astonishing proportions: in the 19th century alone, Tsimshian numbers fell by 57 percent. A major focus of Lindo’s paper was the period in the wake of this collapse. How did the genomes of the Tsimshian respond to this traumatic evolutionary event? What Lindo found was that, in terms of the variety in their genomes, the Tsimshian rebounded surprisingly well. “We didn’t see a decrease in genetic diversity,” he says, “which would have been bad for fighting off diseases and things like that.” Rather, the Tsimshian population maintained the crucial genetic diversity that any population needs to survive. “It seems to be because, after European contact, this particular people started intermarrying with others,” Lindo says, “which likely wasn’t the case beforehand. And intermarrying with immigrants as well.” This was an important factor in keeping their population genetically resilient. “It increased genetic diversity,” he says, “which mitigated to a certain extent the negative effects of the collapse.” From the first stages of his research, Lindo was in direct contact with cultural ambassadors of the Tsimshian community, who advised his team on how to respectfully present its findings and received co-authorship credit for their input. “They reviewed the paper before we submitted it,” Lindo says, “to make sure the wording was sensitive to their culture and their whole histories.” One key takeaway from the Tsimshian reviewers’ feedback was that speculative “storytelling” was to be avoided in the paper. Where Lindo and his team don’t know something—such as precisely why the population experienced a long slow decline—they admit it rather than invent a narrative. Lindo is hopeful that the Tsimshian people more broadly will find value in the new research. “After European colonization, there was a big disruption in their culture, and in transmitting their oral histories from one generation to the next," he said. "And this might help them connect to their ancient history before European contact a little better.” |
|
|
|
Post by Admin on May 11, 2020 7:25:10 GMT
Patterns of Genetic Coding Variation in a Native American Population before and after European Contact John Lindo Mary Rogers Elizabeth K. Mallott Jerome S. Cybulski Ripan S. Malhi Michael DeGiorgio The effects of European colonization on the genomes of Native Americans may have produced excesses of potentially deleterious features, mainly due to the severe reductions in population size and corresponding losses of genetic diversity. This assumption, however, neither considers actual genomic patterns that existed before colonization nor does it adequately capture the effects of admixture. In this study, we analyze the whole-exome sequences of modern and ancient individuals from a Northwest Coast First Nation, with a demographic history similar to other indigenous populations from the Americas. We show that in approximately ten generations from initial European contact, the modern individuals exhibit reduced levels of novel and low-frequency variants, a lower proportion of potentially deleterious alleles, and decreased heterozygosity when compared to their ancestors. This pattern can be explained by a dramatic population decline, resulting in the loss of potentially damaging low-frequency variants, and subsequent admixture. We also find evidence that the indigenous population was on a steady decline in effective population size for several thousand years before contact, which emphasizes regional demography over the common conception of a uniform expansion after entry into the Americas. This study examines the genomic consequences of colonialism on an indigenous group and describes the continuing role of gene flow among modern populations.  Figure 1 Admixture Signal between the Modern Tsimshian and Europeans Introduction The indigenous peoples of the Americas suffered extensive population declines associated with the impact of European colonization. Although the precise extent of this decline is contested1, 2 and likely varied with local circumstance,3 these events should have affected the genetic variation within surviving indigenous populations. These effects are, however, further complicated by patterns of gene flow from both native and non-native immigrant groups. Although previous studies have explored the genetic diversity of contemporary Native American populations,4, 5, 6, 7, 8 the effects of colonization have not been examined with the aid of studies concerning ancient Native American genetic (autosomal) diversity. Here we examine the effects of colonization by comparing the genome-wide patterns of an indigenous population from two different time frames: before and after European contact. Broadly speaking, genetic variation among human populations can result from numerous sources, including stochastic (i.e., mutation, recombination, migration, and genetic drift) and deterministic (i.e., natural selection) processes.9 With the advent of cost-effective genome-wide sequencing, it has become easier to study genetic patterns across the genome in many individuals simultaneously. Statistical analyses of large datasets can be used to reconstruct key events in human evolutionary history, which are discernable from the distribution of allele frequencies across global populations.10, 11, 12 These events include the widely accepted out-of-Africa dispersal, as well as numerous founder effects and population expansions that subsequently occurred as early humans spread throughout the globe.13, 14 Inferring the demographic history of indigenous populations in the Americas has proven difficult due in part to its multi-faceted nature—which likely involved a combination of founder effects, population size changes, and recent admixture.5, 8, 15 The short evolutionary timescale of the effects caused by European colonization further complicate the picture since most of the population-level genomic patterns previously identified involve much longer periods of time, spanning thousands instead of hundreds of years.16, 17, 18 Many of the statistical methods used to identify these patterns are, accordingly, best suited to identify demographic patterns that emerge over longer periods of evolutionary time. Hence, while early Native American migrations have been explored,5, 19 as have the admixture effects of European colonization,8, 20, 21 these recent admixture events have not yet been studied with the aid of comprehensive data concerning patterns of ancient genomic diversity in these populations. In this study, we compare the genomic patterns, offered by previously published whole-exome sequence data,22 of an ancient indigenous population from the Americas (i.e., before any effects of European contact) with the genomic patterns of their modern descendants, the Coast Tsimshian (henceforth, “Tsimshian”). The Tsimshian have occupied Prince Rupert Harbour, British Columbia, since at least 6,000 years before present (BP), as attested to by oral traditions, archeological context, and genetic evidence.22, 23, 24 Similar to other indigenous groups of the Americas, the Tsimshian suffered dramatic population declines in response to the effects of European contact, with a culmination of smallpox epidemics in the 1800s.25 The population collapse likely occurred roughly 175 years ago, with a 57% reduction in effective population size.22 After the epidemics, the Tsimshian intermarried with non-Natives, likely individuals of European descent.8, 22 Given the severity of the Tsimshian population collapse, the expectation would be to find a reduction in overall fitness between the modern and ancestral groups, with some evidence of this at both the individual and population levels.26 This expectation is due to the likely effects of population collapses, which lead to decreases in expected heterozygosity due to reductions in effective population size.27, 28 Through our comparison, however, we identify changes in genomic patterns that have resulted from multiple demographic processes and paint a more nuanced picture, which include the effects of admixture8, 22 (Figure 1). Our study offers an enriched understanding of the genomic impact of the specific demographic factors experienced by the indigenous populations of the Americas. |
|
|
|
Post by Admin on May 11, 2020 20:59:06 GMT
Results Through the analysis of whole-exome data (Table 1), we set out to directly investigate how the patterns of genetic variation in a Native American population have been altered before and after European contact. Using previously published data22 from the Tsimshian of Prince Rupert Harbour (British Columbia, Canada), we detected 59,912 high-confidence single-nucleotide polymorphisms (SNPs) from 24 modern individuals (post-European contact) and 62,906 from 24 of the Prince Rupert Harbour and Lucy Island ancient individuals (henceforth, “ancients”) (pre-European contact), derived from 62 megabases (Mb) of targeted regions. Table 1 Pre-European Contact Ancient Samples Sample Location Method Coding Read Depth Analyses 125 PRH exome 4 all 158 PRH exome 10 all 163 PRH exome 5.6 all 167 PRH exome 4.8 all 168 PRH exome 28 all 181 PRH exome 27 all 300 PRH exome 7.3 all 302 PRH exome 90 all 311 PRH exome 7.4 all 318 PRH exome 6.3 all 322 PRH exome 6.8 all 357 PRH exome 27 all 365 PRH exome 32 all 386 PRH exome 3.5 all 406 PRH exome 5.2 all 413 PRH exome 17.7 all 443 PRH exome 76 all 468 PRH exome 38.9 all 470 PRH exome 33.6 all 507 PRH exome 7 all 516 PRH exome 15 all 525 PRH exome 5.7 all 532 PRH exome 10.4 all 939 Lucy exome 8.5 all 938a Lucy shotgun 1.1b heterozygosity Shuká Káaa Alaska shotgun 2.7b heterozygosity Abbreviations: PRH, Prince Rupert Harbour, British Columbia; Lucy, Lucy Island, several miles off of the coast of Prince Rupert Harbour. Shuká Káa was found on Prince Edward Island, Alaska. a Genotypes not called due to low coverage and samples used only for heterozygosity analysis. Samples range in age from 10,000 to 1,500 years BP. b Coverage calculated over the exome. Compared with the modern individuals, the ancient group exhibits higher levels of mean observed heterozygosity within coding regions (mean heterozygosity across modern 1.230 × 10−4 versus ancient 4.935 × 10−4 individuals) (Table 2). This observation is consistent with expectations for a population that has experienced a recent and dramatic population collapse. We also used heterozygosity (see Material and Methods) as a proxy to estimate changes in effective population size through time (Figure 3). This particular analysis includes the 10,300-year-old Shuká Káa individual from Prince of Wales Island, Alaska, who was found to have a close genetic affinity to the Tsimshian.30 We also included a 5,670-year-old individual, 938, from Lucy Island, off the coast of Prince Rupert Harbour, who was also found to have a close genetic affinity to the Tsimshian.24 Although both samples underwent shotgun sequencing, only regions overlapping with the exome were utilized for consistency with the exome capture data from the 48 Tsimshian and ancient individuals (Table 1). We observe a significant correlation (Figure 3) between increasing heterozygosity and increasing time BP (Pearson correlation p < 9.95 × 10−11, with p value averaged across all possible samplings of one modern Tsimshian individual). This trend may reflect the population collapse after European contact and not a general trend of effective population decline before contact, which has been observed in studies of indigenous populations of the Americas utilizing mitochondrial DNA.2, 42 Furthermore, when the oldest individual, Shuká Káa, is removed, significance is also maintained (Pearson correlation p < 1.9 × 10−6, with p value averaged across all possible samplings of one modern Tsimshian individual). We also masked the modern population for European ancestry but still found a significant increasing trend in heterozygosity (Pearson correlation with p < 1.21 × 10−9 including and p < 1.51 × 10−5 excluding Shuká Káa, and with p value averaged across all possible samplings of one modern Tsimshian individual). Moreover, rather than calling genotypes, we also considered accounting for uncertainty in genotype calling for the 48 Tsimshian and ancient samples, using the identical pipeline as used for 938 and Shuká Káa. The analyses still maintained a significant increasing trend in heterozygosity (Pearson correlation with p < 2.1 × 10−8 including and p < 5.61 × 10−3 excluding Shuká Káa, and with p value averaged across all possible samplings of one modern Tsimshian individual). There is also variation in heterozygosity across the ancient groups within archaeological sites, which may suggest local demographic factors at play. The modern individuals similarly show variation in heterozygosity (in both masked and non-masked analyses), which may correlate to varying levels of gene flow from other indigenous and non-native populations.8 Table 2 Genetic Measures for the 62 Mb Targeted Regions Individuals Total SNPs Coding SNPs Mean Depth (coding only) Mean Heterozygosity Tajima’s D Ts/Tv Modern 24 59,912 40,311 18.72 1.230 × 10−4 0.471 2.18 Ancient 24 62,906 49,630 18.65 4.935 × 10−4 −0.216 2.42 Depth was calculated per individual, using coding sites, and then averaged across individuals. Heterozygosity was measured per individual at all high-confidence sites, and then averaged across individuals. Tajima’s D was measured on an average across 10 kb windows, with a minimum of 5 segregating sites in each window. Ts/Tv designates the transition/transversion ratio within coding regions; a ratio above 2 is expected.61  Figure 3 Estimated Heterozygosity through Time To maximize the data available within the ancient population for the analyses described below, while safeguarding against DNA damage, which could artificially increase the proportion of low-frequency variants in the ancient individuals, we employed a genotype calling method that specifically considers deamination patterns when calling bases (see Material and Methods). In addition to various filters described in the methods, we also removed singletons to prevent any further biases introduced from deamination in the ancient samples, as this phenomenon is random and likely segregates at very low frequencies at any given site. The same filters and calling method were applied to the modern group to prevent batch effect differences. Furthermore, we included only the 48 exome capture samples in the preceding analyses for consistency (Table 1). Next, we examined the derived site frequency spectrum (SFS; Figure 4). The ancient individuals display a significant increase over the modern in variants with a minor allele frequency (MAF) below 0.05 (z-test, p < 10−4). This result is likely due to two demographic factors: (1) a population expansion after the founder effect from the initial peopling of the Americas in the ancient group and (2) the population collapse after European contact, resulting in the loss of low-frequency variants in the modern group. Further evidence for this early expansion with later contraction arises from the ancient individuals exhibiting a lower Tajima’s D value than the modern individuals (z-test, p = 1.73 × 10−6; Table 2), where negative values (ancient) could be indicative of a population expansion after a bottleneck or founder effect and positive values (modern) could be indicative of a sudden population collapse or development of population structure.43 We also observe an increase in intermediate frequencies in the modern group over the ancient group, which is likely the effect of low-frequency alleles becoming less abundant than alleles at intermediate frequencies shortly after a population collapse.44 |
|
|
|
Post by Admin on May 12, 2020 5:15:03 GMT
 Figure 4 Derived Allele Frequency Spectra Of the combined SNPs, only a small fraction of functional SNPs is shared between the ancient and modern groups (17.4%), which are enriched in the ancient group for derived alleles with frequencies of less than 0.1 (73% of total potentially functional variants). Similar fractions were identified in a French-Canadian population using an analogous approach.45 We also confirmed that the C→T and G→A transitions attributable to ancient DNA damage, which may not have been accounted for by the deamination-based calling method, were not driving the differences between the two groups (see Material and Methods and Figure 2). However, despite seemingly disparate features between the two sampling time frames (i.e., before and after European contact), genome-wide FST between the ancient and modern samples remains relatively low (FST = 0.022). Because the differences in low-frequency functional SNPs between the two groups are considerable, we discuss the effects of these variants. We do this, first, by testing for differences in the ratio of nonsynonymous to synonymous changes as a function of minor allele frequency (Figure 5). The nonsynonymous to synonymous ratio of 1.57 in the ancient individuals, for SNPs with a frequency below 0.05, points to a major fraction of potentially deleterious SNPs. The same ratio in the modern is 1.6, which is an increase but not a significant one (p > 0.1, chi-square test). These ratios are higher than those seen in non-Native American populations12, 46 but similar trends are also seen in other studies of deleterious genomic features, such as runs of homozygosity.47, 48 Significant differences between the two groups are found between frequencies of 0.05 and 0.15, where the ancient individuals display a higher ratio compared to the modern individuals (p < 1 × 10−5, chi-square test) (Figure 5A). For the most common variants (above a frequency of 0.25), the two groups again show a non-significant difference between ratios, 1.1 for the ancient and 1.06 for the modern (p > 0.09, chi-square test).  Figure 5 Excess of Potentially Functional Variants in the Ancient Individuals We also tested for the effects of admixture in the modern group by masking European ancestry. The estimated European admixture fraction in the modern group is approximately 30%.8, 22 With the masking, the difference in the ratio for low-frequency alleles becomes significant (p < 6 × 10−3, chi-square test), with the modern showing a marked increase over the ancient at 1.72 versus 1.57, respectively. Three other frequency bins also show increases of the masked modern over the ancient, with variants between frequencies 0.30 and 0.35 showing a significant difference (p < 0.01, chi-square test). Second, because previous studies predict that low-frequency nonsynonymous variants tend to be deleterious,11, 14, 49 we consider whether this difference in nonsynonymous variants correlate to a shift in the burden of potentially damaging alleles. To assess this possibility, we examine the predicted effects of nonsynonymous variants using the Combined Annotation Dependent Depletion (CADD) scaled scores.50 CADD is a powerful method because it integrates various forms of information, which include conservation, allelic diversity, pathogenicity, and experimentally measured regulatory effects. Scaled scores range from 1 to 99 and are based on the rank of each variant compared to the 8.6 billion mutational positions in the human reference genome (hg19). Scores higher than 10 represent the top 10%, higher than 20 the top 1%, and higher than 30 the top 0.1%, all with respect to potential deleteriousness. Using this combined measure, the ancient individuals demonstrate evidence for an excess of potentially damaging mutations below a frequency of 0.2, when compared with the modern individuals (Figure 5B). This trend is affected by masking European admixture in the modern group, where the masked individuals exhibit higher means than the unmasked and overtake the ancient group on several frequency bins above 0.15 (Figure 5B). Examining only functional sites, the ancient individuals exhibit a larger proportion of scores above 20 than the modern individuals (Figure 6). However, when the modern group is masked for European ancestry, the modern group exhibits a larger proportion of scores above 20, which approaches that of the ancient group. This may indicate that admixture has contributed alleles to the population that are potentially less damaging.  Figure 6 CADD Classification of Functional SNPs in Each Population Third, we examine the number of novel variants in both the ancient and modern individuals. We define novel variants as those that are not found in the populations represented in the 1000 Genomes Project phase 3 release.36 The ancient individuals show an excess of novel alleles when compared with the modern individuals (Figure 7A). This enrichment is likely due in part to the overall loss of genetic variation in the modern individuals caused by the population collapse associated with European colonization, as well as subsequent admixture with non-indigenous populations. The modern individuals exhibit, however, a significant decrease in the portion of the most likely damaging sites within these novel alleles (above the top 0.1%), when compared with the ancient individuals (Figure 7B; modern fraction 0.091, ancient fraction 0.103, z-test p < 9 × 10−3). Previous studies have found that rare and novel alleles tend to be of a deleterious nature11, 51, 52 and an expansion following bottlenecks or founder effects may create a proportionally greater number of these variants in a population. However, these time frames are on the order of thousands of years and it has been less than 150 (only several generations) since the modern group’s population collapse from the effects of European contact.53  Figure 7 Novel Potentially Functional Variants Discussion The impact of European colonization has altered the genomes of Native Americans in multiple and dynamic ways. The data discussed in this study suggest that within approximately nine generations since the time of European contact, the modern group have significantly fewer low-frequency and potentially damaging alleles than their ancient ancestors. The differences between the sampling periods can be partially explained by the population expansion that increased the number of low-frequency alleles in the ancient individuals, following the initial peopling of the Americas.54 Although the genomic signatures of a human population expansion following a founder effect have been explored in other populations,11, 12, 55 the genomic consequences of some more recent and severe demographic events seems to carry additional impacts. The modern individuals have experienced a relatively recent and severe population decline after European contact. This may partially explain the loss of low-frequency and novel alleles when contrasted with the ancient individuals and, in turn, the decrease in the number of potentially deleterious alleles. The effects of such a recent collapse, coupled with slow recovery, is, however, typically expected to amplify certain alleles to higher frequency within a population, due to the distortion of allele frequencies by genetic drift.44 But these expectations are not borne out in our data as a result of two primary factors: first, the relatively short evolutionary timescale within which these events occurred; and, second, the recent admixture with both indigenous and non-indigenous populations, which may have increased genetic diversity and countered the deleterious effects of reduced population size.56 Even at very low levels, gene flow from an admixture event has been observed to increase genetic diversity stemming from the connectivity between two populations.57, 58 The Coast Tsimshian may have established this increased variation through population connectivity, from admixture with both native and non-native groups,8, 59 and we believe recent and rapid genomic changes like these need further study in a broader range of cases. Population collapses can also have a different type of effect in terms of the expected heterozygosity of a population, which is typically expected to decrease due to the associated loss of alleles.44 This decrease is substantially influenced by the magnitude of the population collapse. Despite the relatively short amount of time, the modern individuals show variance in the associated heterozygosity, some reaching the level of their ancestors (Figure 3). However, European admixture does not seem to be a factor here, as variation is seen in both masked and non-masked individuals and is in line with the variation seen in the ancient population through time. It should also be noted that the term “deleterious” to describe the potential consequence of an allele is problematic when discussing a population due to the term’s lack of sensitivity. Here we assess the potential damaging effects of an allele in various ways, including the strength of conservation and the probability that the protein function itself will be altered. These predictions are, however, mainly a tool used by biomedical research to identify a putative disease-causing variant in a population context, which are then explored further via other approaches. Given the uncertainty that any allele marked as “deleterious” has a disease outcome, we utilized the most extreme predictions (i.e., those ranked above the 99th percentile) to examine changes in genomic patterns between two time frames. We do not conclude that ancient Native Americans harbored large reservoirs of deleterious alleles but instead demonstrate a fluctuation of a particular class of alleles through time. The role of novel variants in a population is largely environment dependent and a negative categorization, especially related to the ancient past, is misleading. Native American evolutionary history is complex and involves a maelstrom of demographic processes. Some of these complexities are reflected in the genomic patterns observed here in a single Native American population, before and after European contact. Our study suggests that while the ancient individuals exhibit the trademark genomic patterns of a rapid population expansion following a founder effect, their modern descendants have more nuanced genetic patterns. Although the effects of the recent population collapse, associated with European contact, seem to have removed a large portion of low-frequency alleles from the population, this reduction is not accompanied by the same expected increase of potentially deleterious genomic features. For reasons discussed, this pattern may represent the ameliorating effects of allele introgression caused by admixture. We also find a trend of population decline in the ancient group before contact, which could be related to substructure after populations were established regionally in North America. It is also possible that instead of a steady expansion after the entry into the Americas, population size varied by region and was potentially linked to the many cultures and environments of the Americas. These factors could have contributed to the steady population decline in this particular region before European contact. In conclusion, with the use of ancient DNA, we uncovered the genomic patterns of a population representative of the indigenous peoples of the Americas and show how the effects of gene flow have reshaped these patterns in subtle ways. As human populations continue to intermingle through the effects of globalization, this study highlights the impacts of increased gene flow on the genomic patterns between populations with both similar and divergent evolutionary histories. |
|
|
|
Post by Admin on May 20, 2020 19:12:59 GMT
Modern humans have lived near Lake Baikal since the Upper Paleolithic, and have left behind a rich archaeological record. Ancient genomes from the region have revealed multiple genetic turnovers and admixture events, indicating that the transition from the Neolithic to the Bronze Age was facilitated by human mobility and complex cultural interactions. The nature and timing of these interactions, however, remains largely unknown.  A new study published in the journal Cell reports the findings of 19 newly sequenced ancient human genomes from the region of Lake Baikal, including one of the oldest reported from that region. Led by the Department of Archaeogenetics at the Max Planck Institute for the Science of Human History, the study illuminates the population history of the region, revealing deep connections with the First Peoples of the Americas, dating as far back as the Upper Paleolithic period, as well as connectivity across Eurasia during the Early Bronze Age. The deepest link between peoples "This study reveals the deepest link between Upper Paleolithic Siberians and First Americans," says He Yu, first author of the study. "We believe this could shed light on future studies about Native American population history."  Past studies have indicated a connection between Siberian and American populations, but a 14,000-year-old individual analysed in this study is the oldest to carry the mixed ancestry present in Native Americans. Using an extremely fragmented tooth excavated in 1962 at the Ust-Kyahta-3 site, researchers generated a shotgun-sequenced genome enabled by cutting edge techniques in molecular biology. This individual from southern Siberia, along with a younger Mesolithic one from northeastern Sibe-ria, shares the same genetic mixture of Ancient North Eurasian (ANE) and Northeast Asian (NEA) ancestry found in Native Americans, and suggests that the ancestry which later gave rise to Native Americans in North- and South America was much more widely distributed than previously assumed. Evidence suggests that this population experienced frequent genetic contacts with NEA populations, resulting in varying admixture proportions across time and space. "The Upper Paleolithic genome will provide a legacy to study human genetic history in the future," says Cosimo Posth, a senior author of the paper. Further genetic evidence from Upper Paleolithic Siberian groups is necessary to determine when and where the ancestral gene pool of Native Ameri-cans came together. player.vimeo.com/video/419849638A web of prehistoric connections In addition to this transcontinental connection, the study presents connectivity within Eurasia as evidenced in both human and pathogen genomes as well as stable isotope analysis. Combining these lines of evidence, the researchers were able to produce a detailed description of the population histo-ry in the Lake Baikal region. The presence of Eastern European steppe-related ancestry is evidence of contact between southern Siberian and western Eurasian steppe populations in the preamble to the Early Bronze Age, an era characterized by increasing social and technological complexity. The surprising presence of Yersinia pestis, the plague-causing pathogen, points to further wide-ranging contacts. 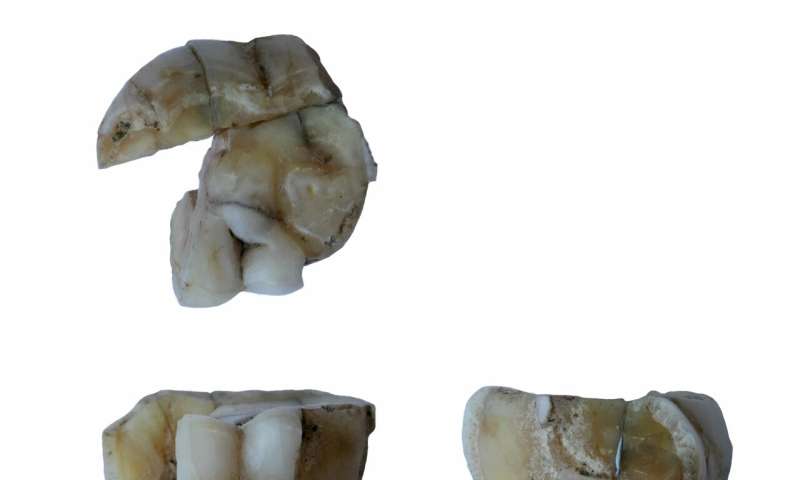 Although spreading of Y. pestis was postulated to be facilitated by migrations from the steppe, the two individuals here identified with the pathogen were genetically northeastern Asian-like. Isotope analysis of one of the infected individuals revealed a non-local signal, suggesting origins outside the region of discovery. In addition, the strains of Y. pestis the pair carried is most closely related to a contemporaneous strain identified in an individual from the Baltic region of northeastern Europe, further supporting the high mobility of those Bronze age pathogens and likely also people. "This easternmost appearance of ancient Y. pestis strains is likely suggestive of long-range mobility during the Bronze Age," says Maria Spyrou, one of the study's coauthors. "In the future, with the generation of additional data we hope to delineate the spreading patterns of plague in more detail." concludes Johannes Krause, senior author of the study.  Paleolithic to Bronze Age Siberians Reveal Connections with First Americans and across Eurasia He Yu Maria A. Spyrou Marina Karapetian Cosimo Posth Choongwon Jeong Johannes Krause 8 Published:May 20, 2020 DOI:https://doi.org/10.1016/j.cell.2020.04.037 Highlights •An Upper Paleolithic Siberian shows a deep link with the First Peoples of the Americas •A 10,000-year continuum of Ancient North Eurasian ancestry in the Lake Baikal region •The Neolithic to Bronze Age population formation occurred through prolonged local admixture •Long-range human and Y. pestis mobility across Eurasia during the Early Bronze Age Summary Modern humans have inhabited the Lake Baikal region since the Upper Paleolithic, though the precise history of its peoples over this long time span is still largely unknown. Here, we report genome-wide data from 19 Upper Paleolithic to Early Bronze Age individuals from this Siberian region. An Upper Paleolithic genome shows a direct link with the First Americans by sharing the admixed ancestry that gave rise to all non-Arctic Native Americans. We also demonstrate the formation of Early Neolithic and Bronze Age Baikal populations as the result of prolonged admixture throughout the eighth to sixth millennium BP. Moreover, we detect genetic interactions with western Eurasian steppe populations and reconstruct Ye r sinia pestis genomes from two Early Bronze Age individuals without western Eurasian ancestry. Overall, our study demonstrates the most deeply divergent connection between Upper Paleolithic Siberians and the First Americans and reveals human and pathogen mobility across Eurasia during the Bronze Age. |
|

