|
|
Post by Admin on May 21, 2020 19:02:14 GMT
An international team of researchers has found evidence that suggests wild relatives of domestic goats passed on a gene to their domesticated relatives that boosts their pathogen resistance. In their paper published in the journal Science Advances, the group describes their study of goat genetic history and what they learned from it.  As the researchers note, by providing a steady source of meat and milk, goat domestication played an important role in the advancement of agriculture, and from there, the development of civilizations. But they also note that it is still not clear what sort of genetic changes domestic goats may have undergone that allowed them to become such a successful domesticated animal. In this new effort, the researchers sought to learn more about such changes by studying the genes of both modern and ancient species. The work involved analyzing the genes of 164 modern domestic goats, 24 modern wild goats, 52 ancient goats and four ancient wild goats from different parts of the world. As part of their analysis, they sequenced all the samples and also carried out a demographic analysis of each as a means of isolating genes that were passed between different species. The researchers found that modern domestic goats got a lot of gene segments from wild mountain goats. And one gene in particular, from a West Caucasian tur-like species, appears to have given modern goats an immune boost. The gene transfer was calculated to have occurred approximately 7,200 years ago, and it helped domesticated goats ward off a pathogen that interferes with digestion. The wild goat was believed to have lived in a subtropical environment where it had adapted to the pathogens found there. The researchers report that almost every species of modern goat has the gene—it codes for a protein produced in the lining of the gut. Testing showed that the few modern species that do not have the gene are more susceptible to infestations of nematode worm eggs. The researchers conclude by suggesting that successful domestication of goats appears to have more to do with genetic additions that warded off pathogens in crowded conditions than for increased milk production. The origin of domestication genes in goats Science Advances 20 May 2020: Vol. 6, no. 21, eaaz5216 DOI: 10.1126/sciadv.aaz5216 Abstract Goat domestication was critical for agriculture and civilization, but its underlying genetic changes and selection regimes remain unclear. Here, we analyze the genomes of worldwide domestic goats, wild caprid species, and historical remains, providing evidence of an ancient introgression event from a West Caucasian tur-like species to the ancestor of domestic goats. One introgressed locus with a strong signature of selection harbors the MUC6 gene, which encodes a gastrointestinally secreted mucin. Experiments revealed that the nearly fixed introgressed haplotype confers enhanced immune resistance to gastrointestinal pathogens. Another locus with a strong signal of selection may be related to behavior. The selected alleles at these two loci emerged in domestic goats at least 7200 and 8100 years ago, respectively, and increased to high frequencies concurrent with the expansion of the ubiquitous modern mitochondrial haplogroup A. Tracking these archaeologically cryptic evolutionary transformations provides new insights into the mechanisms of animal domestication. INTRODUCTION Domestication presents an extreme shift of physiological and behavioral stress for free-living animals (1) to a high-density and disease-prone anthropogenic environment, especially for herbivores. The goat (Capra hircus) was one of the first domesticated livestock species, demonstrating remarkable adaptability and versatility (2, 3), and it has been intimately associated with human dispersal (4). Recent studies have identified candidate targets of selection during goat domestication, including loci related to pigmentation, xenobiotic metabolism, and milk production (5, 6); however, the evolutionary dynamics of key genes involved in adaptation during the early phase of domestication remain unclear. Goat domestication is believed to have begun ~11,000 years ago from a mosaic of wild bezoar populations (Capra aegagrus) (2, 6). However, other wild Capra species are widely distributed and many of them can hybridize with domestic goats (7–9). Their contribution to the goat domestication process remains unexplored. Introgression of adaptive variants has been widely recognized as a notable evolutionary phenomenon in humans (10), sheep (11), and cattle (12), and it may be particularly effective for increasing fitness without negative pleiotropic effects as demonstrated in other species (13). Here, we conducted a comprehensive population genomic survey of modern goat populations distributed across the world, six wild Capra species, and previously published ancient goat genomes to investigate temporal shifts in key genetic variants under selection during goat domestication. RESULTS Population structure and origin of domestic goats We sequenced 101 Capra genomes (coverage, 3 to 47 times; average, 12 times), including 88 domestic goats collected from three different continents, one bezoar, one Alpine ibex (Capra ibex), three Siberian ibex (Capra sibirica), three Markhors (Capra falconeri), one West Caucasian tur (Capra caucasica), and four Nubian ibex × domestic goat hybrids (Capra nubiana × C. hircus) (Fig. 1A and text S1). We also sequenced five ancient goat samples at ~0.04 to 13.44 times coverage (Fig. 1A and text S1), including the earliest known Chinese archaeological samples from the Neolithic time period (14). Together with the publicly available genomic sequences for modern goat and bezoar (table S1) (5, 15), as well as ancient genomes from (table S4) (6), we compiled a worldwide collection of 164 modern domestic goats, 52 ancient goats, 24 modern bezoars, and 4 ancient bezoars.  Fig. 1 Geographic distribution and genetic affinities of wild and domestic goats used in this study. A whole-genome neighbor-joining phylogenetic tree reveals that all domestic goats form a monophyletic sister lineage to bezoars (Fig. 1B and fig. S4), confirming that modern domestic goats are the descendants of bezoar-like ancestors (16). The other four wild Capra species (C. ibex, C. sibirica, C. falconeri, and C. caucasica), which are referred to as the ibex-like species (17), fall exclusively in another branch divergent from bezoar-goat (Fig. 1B and fig. S4). Principal components analysis (PCA) shows that the three bezoar populations, which were collected in the Middle East (Fig. 1A) near the domestication center (2, 18), are structured and cluster corresponding to their geographic origins (Fig. 1C). Within domestic goats, PCA and model-based clustering (k = 3) show that Asian goats are genetically distinct from European (EUR) and African (AFR) samples (Fig. 1, D and E). At k = 6, Asian goats further split into two geographic subgroups: Southwest Asia–South Asia (SWA-SAS) and East Asia (EAS) (Fig. 1E). This geographic structure is also supported by TreeMix (fig. S8) and haplotype-based statistics (fig. S9), in agreement with the scenario that the ancestors of present-day domestic goat populations followed distinct dispersal routes along the east-west axis of Afro-Eurasia (Fig. 2A and table S6) (4). This dispersal scenario was further supported by the observation that populations far from the domestication center (non-SWA) had elevated linkage disequilibrium and reduced genetic diversity (fig. S10).  Fig. 2 Gene flow during the early stage of goat domestication and diffusion. (A) Geographic distribution of the wild Capra species and the dispersal routes of domestic goats out of their domestication areas (3, 7). Sampling locations for ibex-like species are shown by squares. (B) Allele sharing between modern bezoars (H3) and domestic goats. (C) Allele sharing between domestic goats (H3) and ibex-like species (H1). See also table S8 for other populations (H3) investigated. Statistically significant results, defined by |Z scores| ≥ 3, are marked with a red asterisk for (B) and (C). (D) A scatter plot of introgressed haplotype frequency in EUR-AFR and Asian goat populations. The red dots represent immune-related loci. (E) A heat map of D statistic testing for the differential affinity between SWA (blue) and H2 (red), where H2 represents the individual or population of bezoars and domestic goats located in the map. The East Asian goats are depicted in the box in the upper right corner. (F) Haplotype network from the number of pairwise differences at the MUC6 nonrepeat region. All domestic goats are colored in green, and wild Capra species are divided into five subgroups, including the haplotypes from hybrids of Nubian ibex × domestic goat. The radius of the pie chart and the width of edges were log2-transformed. Our demographic analyses using multiple sequentially Markovian coalescent (MSMC), SMC++, and ∂a∂i indicate that the divergence times between modern Asian and European goat populations predated the archaeologically estimated domestication time (~11,000 years ago) (fig. S12 and text S2). In addition, D statistics revealed that the bezoars from Zagros display higher genetic affinity to eastern domestic populations, whereas the bezoars in Azerbaijan show higher affinity to western domestic populations (Fig. 2B and fig. S15A). Therefore, the deep coalescence times found between east-west population pairs can be explained by their origin in structured ancestral bezoar populations (6) or by postdomestication recruitment from different local bezoar populations. |
|
|
|
Post by Admin on May 22, 2020 5:43:16 GMT
Gene flow from ibex-like species to predomestication bezoars and modern goats D statistics reveal that all four ibex-like species have significant signals of allele sharing with ancient and modern goats, indicative of admixture (Fig. 2C and table S8). We then examined this genome-wide pattern of admixture between ibex-like species and domestic goats using D statistics and identity by state in 20-kb sliding windows. We further verified candidate introgressed regions using Sprime and maximum likelihood (ML) phylogenetic trees. Using a conservative criterion (namely, only keeping putative introgressed haplotypes with a frequency higher than 0.1 in goats), we identified 112 genomic segments overlapping with 81 protein-coding genes with signatures of introgression from ibex-like species (Fig. 2D, fig. S16, and data file S1). A Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis for these genes shows that the most significantly enriched category is amoebiasis (hypergeometric test, adjusted P < 5.28 × 10−3: table S10), which is related to parasite invasion and immunosuppression, including four genes (SERPINB3, SERPINB4, CD1B, and COL4A4). Three additional genes (BPI, MAN2A1, and CD2AP) are also involved in immune function (19–21). In these segments, we observed a pronounced signature of putatively introgressed alleles from the West Caucasian tur (fig. S17), consistent with this species showing the greatest genome-wide allele sharing with domestic goats (Fig. 2C). While investigating the history of West Caucasian tur admixture, we found that the West Caucasian tur shared more alleles with all four predomesticated bezoars (an Armenian bezoar dating to >47,000 years ago and three Anatolian bezoars dating to ~13,000 years ago) and especially Armenian bezoar, compared to present-day bezoars and domesticates (Fig. 2E). Further f3 statistics in the form of f3 (modern bezoar, West Caucasian tur; target) support gene flow from the West Caucasian tur to ancient Armenian bezoar (table S9). Together with the fact that three predomestication Anatolian bezoars had a tur-like mitochondrial haplotype (6), our results provide evidence for a predomestication admixture event of bezoar with a tur-like species. This ancient admixture event is further supported by TreeMix analyses (fig. S8C) and the close geographical proximity between the West Caucasian tur and the ancient Armenian bezoar, with both being, in turn, closer to the goat domestication center than any other living ibex-like species (Fig. 2A). Although all modern goat genomes suggest admixture with tur, Neolithic Balkan and modern EUR and AFR goats show more allele sharing with tur compared to Asian goats (fig. S15B), probably because of their higher genetic affinity with the ancient Armenian and Anatolian bezoars (Fig. 2E) (6). Therefore, predomestication introgression from a tur-like species may have contributed alleles to ancient bezoars and thereby early domesticated goats and their derived modern populations. Notably, one introgressed region on chromosome 29 has a nearly fixed introgressed haplotype (frequency = 95.7%) in the modern worldwide domestic goats (Fig. 2F and fig. S22A). This region contains one intact protein-coding gene, MUC6, which encodes a gastrointestinally secreted mucin that forms a protective glycoprotein coat involved in host innate immune responses to the invasion of multiple gastrointestinal pathogens (22–24). We searched for potential donor populations in our sequenced wild caprid genomes. The haplotype network constructed with the nonrepeated region of MUC6 shows that the most common domestic haplotype (MUC6D) is similar to West Caucasian tur, differing by only one mutation, while being different from the minor frequency haplotypes of domestic goats and the common haplotype in bezoar (MUC6B) (Fig. 2G). To further validate this introgression signal, we estimated the most recent common ancestor of the divergent haplotypes and investigated whether selection acting on either a de novo mutation or on standing variation could possibly lead to the pattern in this region within domestic goats. Coalescence time estimates (fig. S18A) and neutral simulations (fig. S18B) suggest that the observed pattern of haplotype differentiation is highly unlikely in the absence of interspecific gene flow. Therefore, the nearly fixed MUC6D in goats was most likely introgressed from a lineage close to the West Caucasian tur, consistent with the genome-wide signal. Domestication-mediated selection on immune and neural genes To identify key selective sweeps in goat domestication, we compared the worldwide domestic goat populations with all 24 bezoars by estimating pairwise genetic differentiation (FST), differences in nucleotide diversity (π ln-ratio bezoar/domestic), and cross-population extended haplotype homozygosity (XP-EHH) in 50-kb sliding windows along the genome (figs. S19 and S20). We defined the windows with significant values (Z test, P < 0.005) in all three statistics (FST > 0.195, π ln-ratio > 0.395, and XP-EHH > 2.10) as putative selective sweeps. After merging consecutive outlier windows, 105 unique regions containing 403 protein-coding genes were identified (Fig. 3A and data file S2). Eighteen of these regions have been identified in previous domestication scans, including those associated to phenotypic effects related to immunity, neural pathways or processes, pigmentation, and productivity traits associated with milk composition and hair characteristics (data file S2). The modest overlap with previous studies is mainly due to differences in sample sets, selection scan methodology, and different versions of the reference genome (text S3).  Fig. 3 Genomic regions with selection signals in domestic goats. (A) Distribution of the pairwise fixation index (FST) (x axis), π ln ratio (y axis) and value of XP-EHH (color) between bezoars and domestic goats. The dashed vertical and horizontal lines indicate the significance threshold (corresponding to Z test, P < 0.005, where FST > 0.195, π ln ratio > 0.395, and XP-EHH > 2.1) used for extracting outliers. (B) KEGG pathways identified as significantly overrepresented (hypergeometric test, adjusted P < 0.01). (C and D) Selective sweep and distribution of the recombination (RHO) on chromosome 29 (46.22 to 46.31 Mb) (C) and selective sweep on chromosome 15 (32.24 to 32.37 Mb) (D). The putative sweep region is additionally validated by FST, Tajima’s D, and CLR test. Horizontal dashed lines represent the whole-genome mean for the corresponding parameters. Gene annotations in the sweep region and SNPs nearly fixed for derived alleles in domestic goats (C and D) are indicated at the bottom. A KEGG analysis showed that 9 of the 14 significantly enriched pathways (hypergeometric test, adjusted P < 0.01) are immune related (hypergeometric test, adjusted P = 5 × 10−3 to 2.35 × 10−7) (Fig. 3B and table S12), including influenza A, malaria, African trypanosomiasis, natural killer cell–mediated cytotoxicity, measles, herpes simplex infection, cytokine–cytokine receptor interaction, hepatitis C, and adipocytokine signaling pathway. We surveyed the literature and identified 40 additional genes with immune function (table S13), obtaining a total of 41 regions that contain 77 immune-related genes. Among these, FST is particularly high in the MUC6 region, and we detected 16 missense single-nucleotide polymorphism (SNP) mutations showing FST > 0.88 in this gene (windowed FST = 0.89, π ln-ratio = 1.01, and XP-EHH = 5.25; Fig. 3C and table S14). Two of the 16 missense mutations were predicted to be deleterious, and these deleterious alleles occur at very low frequency (mean = 0.038) in the domestic goat population (fig. S21). Overall, this region contains 228 SNPs nearly fixed for the derived allele in modern goats (frequency > 95%, absent in bezoars) accounting for 93.8% of such variants in the whole genome (a total of 243, data file S3), illustrating the singular nature of this selection signal. The selection enrichment analysis furthermore pinpoints 12 genes involved in neuroactive ligand–receptor interaction (hypergeometric test, adjusted P = 3.70 × 10−5). We observed an additional 37 genes linked to other functions in the nervous system that were under selection during goat domestication (table S15). One genomic region located on chromosome 15 shows the strongest combined signal of selection (FST = 0.90, π ln-ratio = 3.20, and XP-EHH = 8.09) (Fig. 3A). Evidence for large negative Tajima’s D values, high composite likelihood ratio (CLR) scores, and extensive haplotype sharing in domestic goats all suggest strong positive selection at this locus (Fig. 3D and fig. S22B). This locus contains 15 SNPs almost fixed for the derived allele in domestic goats and harbors two protein-coding genes, STIM1 and RRM1 (data file S3). STIM1 is an endoplasmic reticulum calcium sensor involved in regulating Ca2+ and metabotropic glutamate receptor signaling in the neural system (25, 26). It is known for its role in modulating the excitability of Purkinje neurons in the cerebellum, which play a key role in motor learning and the integration of sensory-motor information (27). RRM1 encodes ribonucleoside-diphosphate reductase large subunit, an enzyme essential for the production of deoxyribonucleotides, and influences sensitivity to valproic acid–induced neural tube defects in mice (28). Hence, we hypothesize that this strongly selected locus may be related to neural function and/or behavior. |
|
|
|
Post by Admin on May 22, 2020 20:00:27 GMT
Introgressed MUC6 plays a role in pathogen resistance The MUC6 region constitutes the only intersection between the selection and introgression scans. In sheep and cattle, MUC6 is associated with gastrointestinal parasite resistance (29, 30). The results of transcriptome sequencing, quantitative real-time polymerase chain reaction (qPCR), and immunohistochemistry revealed that MUC6 is specifically expressed in the abomasum and duodenum of goat (Fig. 4A and fig. S23). Structurally, MUC6 has a high and variable number of tandem repeats (VNTRs) rich in Pro, Thr, and Ser residues, which can influence the covalent attachment of O-glycans (31). We therefore used PacBio SMRT transcriptome sequencing to investigate the difference between the MUC6D and MUC6B at the transcriptional level. Besides a number of small indels, an 82–amino acid deletion containing three copies of VNTR after the 2789th amino acid was uniquely identified in MUC6D compared to MUC6B (figs. S24 and S25). The different number of VNTRs in these two haplotypes may influence the function of MUC6, which is key to the generation of gastrointestinal mucous gel (23, 32). To examine the potential difference of pathogen resistance of the MUC6D and MUC6B variants, we carried out an epidemiological survey in a polymorphic goat population. By evaluating fecal egg counts (FEC) for gastrointestinal nematodes in 143 MUC6D/MUC6D, 111 MUC6D/MUC6B, and 14 MUC6B/MUC6B ewes at 13 months of age, we found that the goats carrying MUC6D/MUC6D exhibited lower FEC than those carrying the other two genotypes (Fig. 4B and data file S4), implying that the goats carrying MUC6D might be more resistant to gastrointestinal nematodes. To control for the genetic background, we also sequenced a subset of 117 animals to 5 average depth (data file S4). A genome-wide association analysis for FEC by incorporation of principal component covariates and pairwise relatedness found a strong association signal that contains MUC6 locus on chromosome 29 (42.77 to 51.33 Mb) (Fig. 4C). These results support that the introgressed MUC6D in domestic goats is most likely under selection due to its advantage in the host innate immune response toward potential gastrointestinal pathogens (23).  Fig. 4 The association of MUC6 with gastrointestinal nematodes resistance. (A) Immunohistochemistry for MUC6 in abomasum pyloric and duodenal bulb of a goat. Photomicrographs at 4× and 20× are shown on the left and in the middle. The negative controls (20×) are shown on the right. (B) The statistical association between MUC6 genotype and the FECs for gastrointestinal nematodes. Wilcoxon rank sum test was used to compute the P values. (C) Manhattan plot of genome-wide association results for FECs. The significant SNPs within MUC6 locus are highlighted in dark green. The dotted horizontal line indicates the threshold (−Log10(P) = 7.65). The origin and diffusion of domestic STIM1-RRM1 and MUC6 alleles An in-depth genetic survey of ancient genomes throughout the Near East revealed that two ancient Balkan goats dating to ~8100 years ago carried STIM1-RRM1D (fig. S28 and data file S5). MUC6D appeared later at ~7200 years ago in Southwest Iran (fig. S28 and data file S6), a period characterized by an increased density of settlements in the Fertile Crescent (33). Herding goats at higher densities in a crowded and disease-prone anthropogenic environment would likely have exerted an increased selective pressure for livestock pathogen resistance (34). The first detected ancient animals having STIM1-RRM1D or MUC6D both carry mitochondrial haplogroup A, although this mitochondrial haplogroup had a low frequency and narrow distribution before 7500 years ago (6). By ~6500 years ago, the frequency of the STIM1-RRM1D and MUC6D increased to ~60% in the Near East (Fig. 5A) and spread to China ~3900 years ago (Fig. 5B), concomitant with the consolidation and diffusion of livestock-based economies throughout Eurasia (35, 36). The expansion of these two selected loci was also contemporaneous with the overwhelming spread of mitochondrial haplogroup A. In contrast, the frequencies of the two major Y-chromosome haplogroups remained relatively unchanged over time (Fig. 5B).  Fig. 5 The emergence and diffusion of domestic STIM1-RRM1 and MUC6 haplotypes are concurrent with the spread of mtDNA haplogroup A. (A) The temporal changes in the frequency of the STIM1-RRM1D, MUC6D, and mtDNA haplogroup A from predomestication bezoars to modern domestic goats. The dates are expressed as cal. BP. (B) Genotypes of STIM1-RRM1 and MUC6, and mtDNA and Y-chromosome haplogroups. The presences in homozygous or heterozygous states are shown in green and light green, respectively. The absence of the domestic allele is depicted in light pink. The light gray color symbolizes missing information. DISCUSSION The present study generated genomic data from a substantial number of domestic and wild Capra species to characterize adaptive introgression and genetic changes during goat domestication. Collectively, both the selective sweep enrichment analyses and the two outstanding selection signals found in MUC6 and STIM1-RRM1 in domestic goats are consistent with the hypothesis that genes related to pathogen resistance (37) and behavior have been particularity targeted during goat domestication. Several studies have shown that adaptive introgression can provide beneficial alleles that allow the recipient populations to adapt to new environments (12, 38, 39). In humans, immune-related loci acquired substantially advantageous alleles by immigrant modern humans admixing with archaic humans who were probably well adapted to local environments and pathogens through their long exposure to them (40–42). We here report numerous genomic segments putatively introgressed from an ibex-like species. These introgressed segments are enriched in genes with an immune function, suggesting that historic gene flow from wild species played an important role in shaping the diversity of immune system phenotypes in domestic goat and possibly increased its adaptive potential. When comparing the set of putatively introgressed alleles in each segment to our sampled ibex-like genomes, we find that most match rates range from 0.5 to 0.8 (fig. S17), indicating the degree of similarity between the introgressing and our sequenced ibex-like individuals. Additional wild Capra genome sequences in the future may clarify the origin of these alleles, although it is also possible that the introgression came from a now extinct branch on the Capra tree. In particular, a number of lines of evidence support that gene flow between West Caucasian tur and goats occurred before the onset of domestication. The West Caucasian tur is distributed around the Black Sea coast (Fig. 2A), geographically close to the goat domestication center. It inhabits a humid, subtropical environment where the expected pathogen exposure and parasite load may be considerably higher than in more arid regions of Southwest Asia. The rigorous identification of introgression segments in domestic goats shows that the nearly fixed MUC6D was introgressed from a West Caucasian tur-like species. Although the earliest sign of introgression from West Caucasian tur is old (>47,000 years ago), we did not find the MUC6D in ancient and modern sampled bezoars. Two possibilities can explain this observation. First, given the pervasive gene flow between different Capra species (8, 9, 43), we cannot rule out the possibility of multiple introgression from ibex-like species before and after goat domestication. Second, because of the scarcity of ancient and modern bezoars, it may be that the introgressed variants were segregating at low frequencies in these populations. Nonetheless, our results indicate that the introgression of advantageous MUC6D might enhance innate immune reactivity against potential gastrointestinal pathogens under a grazing agro-ecological niche (44) with previously unknown infectious disease pressures. Our dataset including several ancient goat samples allowed us to tentatively track the emergence and spread of the advantageous variants in these two important domestication loci (Fig. 5). The first occurrences of the STIM1-RRM1D and MUC6D locus coincided with the otherwise rare mitochondrial DNA (mtDNA) haplogroup A, and the three increased in frequency roughly concurrently (Fig. 5A), becoming nearly fixed in modern goat populations. These results suggest that the global diffusion of variations central to the domestication process was possibly mediated by a subset of female goats carrying the mtDNA haplogroup A. Despite this, modern goat populations remain differentiated from each other. The most likely explanation we can think of is that even Neolithic goat husbandry entailed some kind of breeding strategy under which immigrant matrilines containing globally advantageous alleles were deliberately crossbred with local populations, which probably carried local genetic adaptations, rather than simply replacing local populations. Overall, our results provide evidence for adaptive introgression and the genetic basis of selected traits during the domestication of goats. We highlight that livestock domestication is a dynamic evolutionary process, with adaptive leaps driven by introgression and selection. Our results indicate that domestication may have profound impacts on neural traits and pathogen resistance, which helped manage herds to adapt to an anthropogenic environment. |
|
|
|
Post by Admin on Sept 18, 2020 19:24:13 GMT
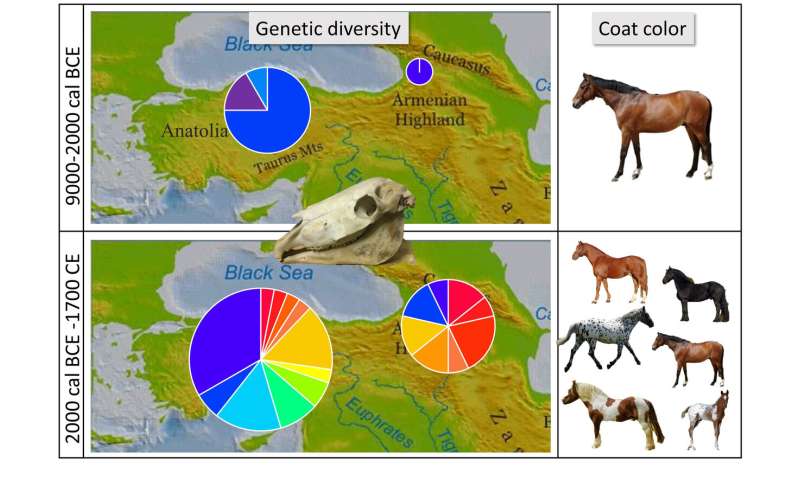 An international team of researchers has found via genetic testing that horse domestication very likely did not begin in Anatolia as has been thought. Instead, it appears more likely that horses were first domesticated in the Eurasian Steppe and were subsequently imported to both the Caucasus and Anatolia. In their paper published in the journal Science Advances, the group describes their exhaustive study of ancient horse remains from a host of locations in eastern parts of Asia, the Caucasus and Anatolia.  For many years, scientists have believed that the horse was first domesticated in Anatolia approximately 5,500 years ago. Anatolia is the peninsula also known as Asia Minor; today it makes up most of Turkey. In this new effort, the researchers have found evidence that suggests that horses were actually first domesticated in the Eurasian Steppe and were exported to Anatolia approximately 4,000 years ago, during the Bronze Age.  The work involved obtaining and genetically analyzing 100 equid remains that had been found at eight sites in Anatolia and six in the Caucasus (a region between the Black Sea and the Caspian Sea that is today mainly occupied by Armenia) dating back 2,500 to 11,000 years ago. Some of the remains were from domesticated horses, while others were from those that had remained wild. To learn more about their origins, the researchers conducted paleogenetic and morphological studies that included analysis of Y chromosome DNA, mitochondrial DNA and DNA markers that have previously been associated with coat color. Over time, domestication has led to changes in the coat color of horses. They found lineages present in modern domestic horses that appeared suddenly in 2,000 BCE horses (as opposed to showing up over time) which suggested that domestication had occurred elsewhere. A sharp change in coat colors also suggested horses had been brought to the region from somewhere else. The researchers suggest that the other location was likely north and west of the Caucasus, closer to the Black Sea—the exact site is still unknown. The researchers also found evidence of imported domesticated horses being bred with wild Anatolian horses, and also donkeys. They found evidence of the earliest known mule in southwest Asia. More information: Silvia Guimaraes et al. Ancient DNA shows domestic horses were introduced in the southern Caucasus and Anatolia during the Bronze Age, Science Advances (2020). DOI: 10.1126/sciadv.abb0030 |
|
|
|
Post by Admin on Sept 19, 2020 0:43:21 GMT
Ancient DNA shows domestic horses were introduced in the southern Caucasus and Anatolia during the Bronze Age
Science Advances 16 Sep 2020:
Vol. 6, no. 38, eabb0030
DOI: 10.1126/sciadv.abb0030
Abstract
Despite the important roles that horses have played in human history, particularly in the spread of languages and cultures, and correspondingly intensive research on this topic, the origin of domestic horses remains elusive. Several domestication centers have been hypothesized, but most of these have been invalidated through recent paleogenetic studies. Anatolia is a region with an extended history of horse exploitation that has been considered a candidate for the origins of domestic horses but has never been subject to detailed investigation. Our paleogenetic study of pre- and protohistoric horses in Anatolia and the Caucasus, based on a diachronic sample from the early Neolithic to the Iron Age (~8000 to ~1000 BCE) that encompasses the presumed transition from wild to domestic horses (4000 to 3000 BCE), shows the rapid and large-scale introduction of domestic horses at the end of the third millennium BCE. Thus, our results argue strongly against autochthonous independent domestication of horses in Anatolia.
INTRODUCTION
The domestication of the horse ca. 5500 years ago represents one of the most important technological innovations in the ancient world (1, 2). With the harnessing of horsepower, political, economic, and social relationships throughout the ancient world were transformed as horses revolutionized transportation and affected patterns of trade, warfare, and migration [e.g., (1–4)]. Archaeological, organic residue, and genetic analyses suggest that the domestic horse originated in the Central Asian steppes, then spread into eastern Europe and later into southwest Asia (SWA) [e.g., (2, 5–11)]. In particular, data from the Botai hunter-gatherer culture in Kazakhstan suggest that, by the mid to late fourth millennium BCE, horses were bitted, milked, selected for the TRPM1 coat color locus, and kept in enclosures and were therefore under intensive management (10, 12–14). A recent study of ancient horse genomes, however, challenged the view that modern domestic horses derived from Central Asia, because “Botai-like” horses were shown to be the ancestors of northeast Asian Przewalski’s horses but not the main source of ancient or modern domestic horses (14, 15). Another recent study ruled out a second potential horse domestication center, the Iberian Peninsula, showing that Iberian wild horses went extinct without leaving notable traces in the genomes of modern horses (16). There are, however, two more areas that have been proposed as domestication centers for modern horses: the Pontic-Caspian steppe (17) and Anatolia (8, 18). Although the former has been long hypothesized as the likely source of domestic horses (12, 13, 19), the latter region has been poorly explored regarding its role in horse domestication processes (20–22), despite its long history of wild horse exploitation and its reputation for breeding valuable horses in Classical Antiquity (23).
The origin of the domestic horse in Anatolia, and more generally in SWA, continues to represent a complex archaeological puzzle. A combination of textual, iconographic, and archaeozoological data suggest that, by the mid to late third millennium BCE, domestic horses were introduced from neighboring mountain regions into Mesopotamia (modern Iraq and northeast Syria), where they were often referred to in cuneiform texts as ANŠE-KURRA (“donkey of the mountain”) (24–28). Initially kept only in small numbers, horses rose to prominence across SWA within a few centuries in association with the spread of chariots, a technological innovation of the second millennium BCE (1, 29). Because horse domestication in the Eurasian steppes, a region historically known for its “horse cultures,” likely began in the fourth or perhaps even fifth millennium BCE (17, 30–32), it has long been argued that southwest Asian horses are the descendants of these early domesticates, which arrived in the region via poorly understood participation in Pontic-Caspian-Transcaucasian interaction spheres or population movements (33, 34).
Another hypothesis argues that Anatolia played a central role in the transmission of domestic horses into Syro-Mesopotamia (33, 35, 36), and an Anatolian contribution to early domestic populations has been suggested (37–40). Archaeological data indicate the widespread presence of wild horses (Equus ferus) and also of so-called hydruntines in early and middle Holocene Anatolia that were regularly exploited (20, 22, 40, 42). The continuity of human-horse interactions from the ninth millennium through the second millennium BCE, when domesticates are known from archaeological contexts, led to the hypothesis that Anatolian wild horses may have been a potential source population for domestic horses. The lack of reliable osteomorphological criteria for differentiating the skeletal remains of wild and domestic horses, however, has hampered attempts to address the hypothesis of a local domestication. Therefore, the cultural processes and mechanisms triggering the widespread appearance of domesticates in the late third millennium are still elusive. In this study, we take advantage of the abundance of archaeological horse remains from the central Anatolian plateau (20, 42–46) to provide the first rigorous test of the hypothesis of Anatolian horse domestication applying paleogenetics.
Complete present-day mitochondrial genomes have revealed 18 major haplogroups (A to R), the radiation times of which date mostly to the Neolithic and later periods (47). In contrast, studies of the mitochondrial hypervariable region in ancient horses have shown that domestic horses exhibit a much higher amount of genetic variation in mitochondrial lineages compared to cattle, sheep, and pigs (48–51). Furthermore, most mitochondrial lineages observed in domestic horses already existed before domestication (52). These analyses of ancient horses did not yield a clear phylogeographic structure that would allow the spatiotemporal origin(s) of horse domestication to be identified. These findings interpreted as suggesting that the mobility of wild horses in northern Eurasia allowed constant population reshuffling and repeated recruitment of wild local mares, precluding the establishment of a phylogeographic structure (52, 53).
In contrast, extant domestic horses exhibit remarkably little variation in the male Y chromosome line, with only one haplotype so far identified in modern domesticates, which led to the early claim of a single domestication event for horses (54, 55). Paleogenomic analyses of ancient specimens, however, observed additional male lineages in prehistoric populations before domestication and revealed that genetically diverse male founders were involved in early domestication (15, 56). This diversity was subsequently reduced, likely as a result of more directed human selection likely starting in the Iron Age and continuing during Roman times (57), and again during the Islamic conquest and the Byzantine-Sassanid war after 7th to 9th c. CE (16).
Paleogenetic evidence from genetic loci associated with coat color in horses argues for a diversification of coat color starting in the Bronze Age and is considered associated with an early stage of the domestication process (11, 58). Because the appearance of new coat colors is common in domestic taxa compared to their wild counterparts, they provide a useful marker for identifying domestic horses in archaeological assemblages.
Up to now, the origins of domestic horses in Anatolia have remained elusive; but careful recovery of horse remains from well-stratified archaeological contexts in Anatolia and in the neighboring Caucasus, together with progress in paleogenetic approaches, now makes it possible to specifically address the processes responsible for the origins of domestic horses in this part of western Asia. For this project, we combined morphological classification of equid remains, which can be hampered by a lack of diagnostic anatomical and/or biometrical criteria (41, 59), with paleogenetic analysis of mitochondrial, Y chromosome DNA, and autosomal DNA markers related to coat color to trace the spatiotemporal dynamics of the emergence of domestic horses in Anatolia. We analyzed over 100 equid remains from 14 prehistoric sites in central Anatolia and the Caucasus covering most of the Holocene (9000 BCE to 1000 CE) to gain insights into the origins of domestic horses in Anatolia, a pivotal issue in near Eastern history.
|
|

