|
|
Post by Admin on Feb 11, 2021 21:29:06 GMT
 Neanderthals' gut microbiota included beneficial microorganisms that are also found in the modern human microbiome. An international research group led by the University of Bologna achieved this result by extracting and analyzing ancient DNA from 50,000-year-old fecal sediments sampled at the archaeological site of El Salt, near Alicante (Spain). Published in Communication Biology, their paper puts forward the hypothesis of the existence of ancestral components of human microbiota that have been living in the human gastrointestinal tract since before the separation between the Homo sapiens and Neanderthals that occurred more than 700,000 years ago. "These results allow us to understand which components of the human gut microbiota are essential for our health, as they are integral elements of our biology also from an evolutionary point of view," explains Marco Candela, the professor of the Department of Pharmacy and Biotechnology of the University of Bologna, who coordinated the study. "Nowadays, there is a progressive reduction of our microbiota diversity due to the context of our modern life: this research group's findings could guide us in devising diet- and lifestyle-tailored solutions to counteract this phenomenon." The issues of the "modern" microbiota The gut microbiota is the collection of trillions of symbiont micro-organisms that populate the gastrointestinal tract. It represents an essential component of human biology and carries out important functions, such as regulating metabolism and the immune system and guarding against pathogenic micro-organisms. Recent studies have shown how some features of modernity—such as the consumption of processed food, drug use, life in hyper-sanitized environments—led to a critical reduction of biodiversity in the gut microbiota. This depletion is mainly due to the loss of a set of microorganisms referred to as "old friends." "The process of depletion of the gut microbiota in modern western urban populations could represent a significant wake-up call," says Simone Rampelli, who is a researcher at the University of Bologna and first author of the study. "This depletion process would become particularly alarming if it involved the loss of those microbiota components that are crucial to our physiology." Indeed, there are some alarming signs. For example, in the West, researchers have noted a dramatic increase in cases of chronic inflammatory diseases such as inflammatory bowel disease, metabolic syndrome, type 2 diabetes and colorectal cancer. How the "ancient" microbiota can help Which components of the gut microbiota are more important for health? Scientists have been seeking targeted solutions. This was the starting point behind the idea of identifying the ancestral traits of human microbiota—specifically, the core of the human gut microbiota, which has remained consistent throughout our evolutionary history. Technology now allows researchers to pursue paleomicrobiology, a new field that studies ancient microorganisms from archaeological remains through DNA sequencing. The research group analyzed ancient DNA samples collected in El Salt (Spain), a site where many Neanderthals lived. To be more precise, they analyzed the ancient DNA extracted from 50,000-year-old sedimentary feces, the oldest sample of fecal material available to date. In this way, they managed to piece together the composition of the micro-organisms populating the intestine of Neanderthals. Comparing the composition of the Neanderthals' microbiota to modern humans revealed many similarities. "Through the analysis of ancient DNA, we were able to isolate a core of microorganisms shared with modern Homo sapiens," explains Silvia Turroni, researcher at the University of Bologna and first author of the study. "This finding allows us to state that these ancient micro-organisms populated the intestine of our species before the separation between Sapiens and Neanderthals, which occurred about 700,000 years ago."  Safeguarding the microbiota These ancestral components of the human gut microbiota include many well-known bacteria that are fundamental to health, among which are Blautia, Dorea, Roseburia, Ruminococcus and Faecalibacterium. By producing short-chain fatty acids from dietary fiber, these bacteria regulate our metabolic and immune balance. There is also the Bifidobacterium, a microorganism playing a key role in regulating immune defenses, especially in early childhood. Finally, in the Neanderthal gut microbiota, researchers identified some of those "old friends." This confirms the researchers' hypotheses about the ancestral nature of these components and their recent depletion in the human gut microbiota due to our modern life context. "In the current modernization scenario, in which there is a progressive reduction of microbiota diversity, this information could guide integrated diet- and lifestyle-tailored strategies to safeguard the micro-organisms that are fundamental to our health," concludes Candela. "To this end, promoting lifestyles that are sustainable for our gut microbiota is of the utmost importance, as it will help maintain the configurations that are compatible with our biology." Explore further Researchers discover link between gut and type 1 diabetes More information: Simone Rampelli et al. Components of a Neanderthal gut microbiome recovered from fecal sediments from El Salt, Communications Biology (2021). DOI: 10.1038/s42003-021-01689-y |
|
|
|
Post by Admin on Feb 12, 2021 5:23:49 GMT
Components of a Neanderthal gut microbiome recovered from fecal sediments from El Salt Communications Biology volume 4, Article number: 169 (2021) Abstract A comprehensive view of our evolutionary history cannot ignore the ancestral features of our gut microbiota. To provide some glimpse into the past, we searched for human gut microbiome components in ancient DNA from 14 archeological sediments spanning four stratigraphic units of El Salt Middle Paleolithic site (Spain), including layers of unit X, which has yielded well-preserved Neanderthal occupation deposits dating around 50 kya. According to our findings, bacterial genera belonging to families known to be part of the modern human gut microbiome are abundantly represented only across unit X samples, showing that well-known beneficial gut commensals, such as Blautia, Dorea, Roseburia, Ruminococcus, Faecalibacterium and Bifidobacterium already populated the intestinal microbiome of Homo since as far back as the last common ancestor between humans and Neanderthals. Introduction Over the past decade, microbiome research has highlighted the crucial role that the gut microbiome plays in human biology through its pleiotropic influence on many physiological functions, such as human development, immunity, metabolism and neurogenerative processes1. This body of knowledge has catalyzed interest in incorporating the gut microbiome into our evolutionary history, as an adaptive partner providing the necessary phenotypic plasticity to buffer dietary and environmental changes. Studies aimed at exploring the ancestral traits of the human gut microbiome are therefore encouraged, as a unique evolutionary perspective to improve our knowledge of gut microbiome assembly and interactions with the human host2. The ancestral configuration of the human gut microbiome has generally been inferred by microbiome data stemming from contemporary populations found across all six human occupied continents who adhere to traditional lifestyles, such as the Hadza hunter-gatherers from Tanzania, the rural Bassa from Nigeria and rural Papuans from Papua New Guinea, among others3,4,5,6,7,8,9,10,11. However, since this research involved modern populations, no direct information on the ancient human gut microbiome structure can actually be provided. Fig. 1: The Middle Paleolithic site of El Salt (Spain).  a, b General site setting. The yellow star marks the location of the excavated area. As can be seen in the photograph, the sedimentary deposit rests against a tall limestone wall. c, d Different views of the excavation area indicating the zones sampled for this study. Zone 1 includes samples V1-3 and Zone 2 all the rest. Alternatively, ancient DNA (aDNA) analysis based on shotgun metagenomic sequencing is emerging as an attractive and reliable opportunity to directly investigate the microbial ecology of our ancestors12,13,14,15. Paleomicrobiological aDNA studies have traditionally been conducted on dental calculus and bones15,16,17,18, providing landmark information on ancient pathogens and oral microbial communities. However, given that stools are widely acknowledged as a proxy of the gut microbiome structure19, the metagenomics of aDNA from paleofeces, also known as coprolites, represents the only way to gain insight into the ancient human gut microbiome2. Pioneering studies in this direction have been carried out, providing next-generation sequencing data from modern human mummified intestinal contents and paleofeces20,21,22,23,24. Nevertheless, to the best of our knowledge, paleofecal samples older than 8,000 years have never being analyzed, leaving an important gap on the pre-historical human gut microbiome configuration. In this scenario, we attempted to identify ancient human gut microbiome components by shotgun metagenomic analysis of aDNA extracted from archeological sedimentary samples (ES1 to ES7) from the stratigraphic unit (SU) X (subunit Xb-H44) of the Middle Paleolithic open-air site, El Salt (Alicante, Spain)25 (Fig. 1). The archeological setting of El Salt yielded evidence of recurrent occupation by Neanderthals, our closest evolutionary relatives, dated between 60.7 ± 8.9 and 45.2 ± 3.4 kya26,27. In particular, the sedimentary samples ES1-7 have been previously shown to include several millimetric phosphatic coprolites and fecal lipid biomarkers, namely coprostanol and 5ß-stigmastanol, with proportions suggesting a human origin25. These samples therefore represent, to our knowledge, the oldest known positive identification of human fecal matter. The present work also includes an additional seven new archeological sediments collected in 2018 as a control. Two were from SU X (subunit Xa and Xb, respectively) and the others from surrounding SUs, i.e., upper V (three samples), IX and XI (one sample each) (Fig. 2a). While SUs IX to XI are associated with rich archaeological assemblages, upper SU V yielded very few archaeological remains26. We found that samples positive for the presence of fecal biomarkers showed traces of both ancient human mtDNA and ancient components of the modern human gut microbiome. These components included so-called “old friends” and beneficial commensal inhabitants of modern human guts, providing unique insights into their relevance to the biology of the Homo lineage.  Fig. 2: Pleistocene stratigraphic sequence of El Salt (Units V-XI), chronometric dates of the sampled units, and evidence of traces of ancient human mitochondrial DNA. a The 14 sediment samples included in this study are shown in red. Samples ES1 to ES7 (subunit Xb-H44) are from Sistiaga et al.25. b Red boxes, human mtDNA fragments as recovered from metagenomic sequencing data; red circles, confirmation of the presence of ancient human mtDNA through target capture and sequencing (please, see Methods for further details). |
|
|
|
Post by Admin on Feb 12, 2021 7:01:24 GMT
Results and discussion Ancient DNA sequencing and damage assessment DNA was extracted from 14 archeological sedimentary samples and prepared for shotgun metagenomics in a dedicated aDNA facility at the Laboratories of Molecular Anthropology and Microbiome Research in Norman (OK, USA) (see Methods). A total of 124,592,506 high-quality paired-end sequences were obtained by Illumina NextSeq sequencing and analyzed for bacterial aDNA. To remove contamination by modern DNA, which is one of the major complications in studies of ancient samples14,28,29, we evaluated the DNA damage pattern as compared with present-day DNA references. In particular, Skoglund and colleagues12 translated the pattern of cytosine deamination into a postmortem degradation score (PMDS), which provides information on whether a given sequence is likely to derive from a degraded aDNA molecule. Reads were aligned against all bacterial genomes of the NCBI database, and ancient bacterial reads were recovered by setting PMDS > 5, to minimize the probability of a sequence being from a present-day contaminating source12. An average of 6,836 sequences per sample (range, 279–17,901) were retained, corresponding to a small but consistent fraction of DNA being ancient and derived from bacteria (mean ± SD, 0.069% ± 0.029%) (Supplementary Table 1 and Supplementary Fig. 1). The same procedure was applied to extraction, library and PCR blanks, resulting in the retrieval of a minimal number of 144, 1, and 42 ancient bacterial sequences, respectively. Ancient reads from blanks were assigned to 116 bacterial species that showed no overlap with the sample dataset (Supplementary Data 1). When comparing the fraction of reads with PMDS > 5 per million reads between samples IX, Xa, ES1-7, Xb and XI (i.e., those positive for the presence of fecal biomarkers and/or associated with rich archaeological assemblages), and samples from SU V (i.e., with no or very few archeological remains), the first showed a greater abundance of PMDS > 5 reads (p-value = 0.01, Wilcoxon test) (Supplementary Fig. 2), possibly as a result of the presence of human fecal sediment. Detection of ancient human mitochondrial DNA In order to detect human aDNA traces in our sample set, we searched for human mitochondrial DNA (mtDNA) sequences in PMDS-filtered metagenomes obtained from the 14 archeological sedimentary samples. Ancient human mtDNA was detected in almost all ES1 to ES7 samples from SU X (Fig. 2b). No traces of mtDNA from other animals were detected. To strengthen these findings, all samples were subjected to target capture of mtDNA with a Neanderthal bait panel (Arbor Biosciences; see Methods), and subsequent sequencing on Illumina NextSeq platform. Based on this analysis, ES1, ES2, ES5 and Xb samples tested positive for the presence of ancient human mtDNA, showing >1000 human mtDNA reads with PMDS > 1, breath of coverage >10%, −Δ % ≥ 0.9 and modern contamination less than 2% (Fig. 2b and Supplementary Table 2)30. Taken together, this evidence strongly supports human origin for the El Salt samples, particularly those from SU X25. Profiling of ancient prokaryotic DNA As for prokaryotic aDNA, seventeen bacterial and one archaeal phyla were identified in the aDNA sedimentary record of El Salt, with different representation across SUs (Fig. 3). As expected for its wide distribution in nature31,32, Actinobacteria is the most represented phylum, with environmental species from Streptomycetaceae, Pseudonocardiaceae, Micromonosporaceae, Nocardiaceae, Mycobacteriaceae, Microbacteriaceae and Nocardioidaceae families being detected in almost all the SUs. Similarly, the vast majority of sediment samples share a number of ancient sequences assigned to Bacillaceae members, which are known to play fundamental roles in soil ecology, where they can persist up to thousands of years, if not longer, due to their ability to form resistant endospores33,34. Another large fraction of aDNA shared by most SUs includes Proteobacteria constituents, especially from Alphaproteobacteria (mainly Rhodobacteraceae, Rhodospirillaceae and Sphingomonadaceae families), Betaproteobacteria (mainly Comamonadaceae and Burkholderiaceae) and Gammaproteobacteria (with Xanthomonadaceae) classes. Again, these are cosmopolitan bacteria commonly found in both terrestrial and aquatic environments, as free-living organisms or symbionts in different hosts35,36. In light of their DNA damage pattern, it is reasonable to assume that these are truly ancient environmental bacteria that populated archeological sediments. The contamination of archeological remains by environmental bacteria is indeed well expected, as already documented in previous paleomicrobiological aDNA studies15,18,37. For the relative abundances of bacterial families detected across the samples, please see Supplementary Data 2. Fig. 3: Ancient bacteria in sediment samples from El Salt. 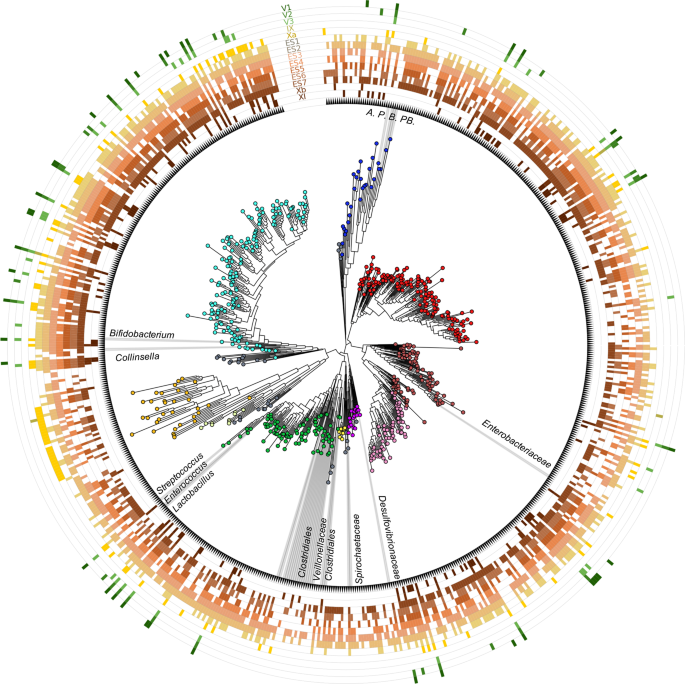 The phylogenetic tree was built with representative sequences of bacterial genera for which at least one species was present with more than four hits in one sample. Different colors indicate different phyla (or classes for Proteobacteria) as follows: cyan, Actinobacteria; blue, Bacteroidetes; red, Alphaprotebacteria; brown, Gammaproteobacteria; pink, Betaproteobacteria; purple, Deltaproteobacteria; yellow, Acidobacteria; green, Firmicutes; light-yellow, Planctomycetes; orange, Euryarchaeota; grey, others (including Armatimonadetes, Chlorobi, Chloroflexi, Cyanobacteria, Deinococcus-Thermus, Fusobacteria, Gemmatimonadetes, Nitrospirae, Spirochaetes, Synergistetes and Verrucomicrobia). Bacterial taxa belonging to families common to the gut microbiome of hominids are highlighted at different taxonomic level. A, Alistipes; P, Prevotella; B, Bacteroides; PB, Parabacteroides. |
|
|
|
Post by Admin on Feb 12, 2021 19:34:10 GMT
Putative components of the Neanderthal gut microbiome Next, following an approach similar to Weyrich et al.18, who first characterized the oral microbiome from Neanderthal dental calculus, we focused our analysis on intestinal microorganisms. Specifically, in order to identify potential ancient human gut microbiome components, we searched for bacterial genera belonging to the 24 families that have recently been indicated as being common to the gut microbiome of hominids (i.e., Methanobacteriaceae, Bifidobacteriaceae, Coriobacteriaceae, Bacteroidaceae, Porphyromonadaceae, Prevotellaceae, Rikenellaceae, Tannerellaceae, Enterococcaceae, Lactobacillaceae, Streptococcaceae, Christensenellaceae, Clostridiaceae, Eubacteriaceae, Lachnospiraceae, Oscillospiraceae, Peptostreptococcaceae, Ruminococcaceae, Erysipelotrichaceae, Veillonellaceae, Desulfovibrionaceae, Succinivibrionaceae, Enterobacteriaceae and Spirochaetaceae)38,39,40,41,42,43,44,45. Accordingly, while harboring similar family-level gut microbiome profiles, humans and non-human hominids, including our closest living relatives—chimpanzees, can be differentiated on the basis of the particular pattern of associated gut microbiome genera (as well as species and strains) represented within these families45. This strong association between gut microbiome composition and host physiology—known as phylosymbiosis—is believed to be universal in mammals, essentially as a result of all the physical, chemical and immunological factors that differentiate the intestine of the host species (e.g., type of digestive organs, pH, oxygen level, host-derived molecules and immune system)46. According to our findings, 210 bacterial species belonging to hominid-associated gut microbiome families, as listed above, are represented in the aDNA from El Salt SU IX, X and XI, with the highest detection rate in samples from SU X and, particularly, in ES1 to ES7 (Fig. 4), for which a human-like host origin had been previously suggested25. In Supplementary Fig. 3, we provide the overall compositional profile of the El Salt samples from SU IX, X and XI restricted to the hominid-associated gut microbiome families, while in Supplementary Fig. 4, the proportions of these families are compared with those of the samples with no or very few archeological remains (i.e., from SU V). The compositional profile of samples from SU IX, X and XI was next compared to publicly available gut microbiomes from contemporary human populations as representative of different subsistence practices, such as Hadza and Matses hunter-gatherers, Tunapuco rural agriculturalists and western urbans from Italy and the US7,47. As shown by the Principal Coordinates Analysis of Bray-Curtis distances between the family-level profiles (Supplementary Fig. 5), the El Salt samples from SU IX, X and XI tend to cluster closer to Tunapuco and Matses, resembling more the “ancestral” human gut microbiome of rural agriculturalists and hunter-gatherers than the urban western gut microbiome7. However, as the degree of degradation of microbial DNA in ancient samples might be different for various gut microbiome components, any conclusions from these compositional data must be taken with due caution. Fig. 4: Ancient components of the human gut microbiome in sediment samples from El Salt. 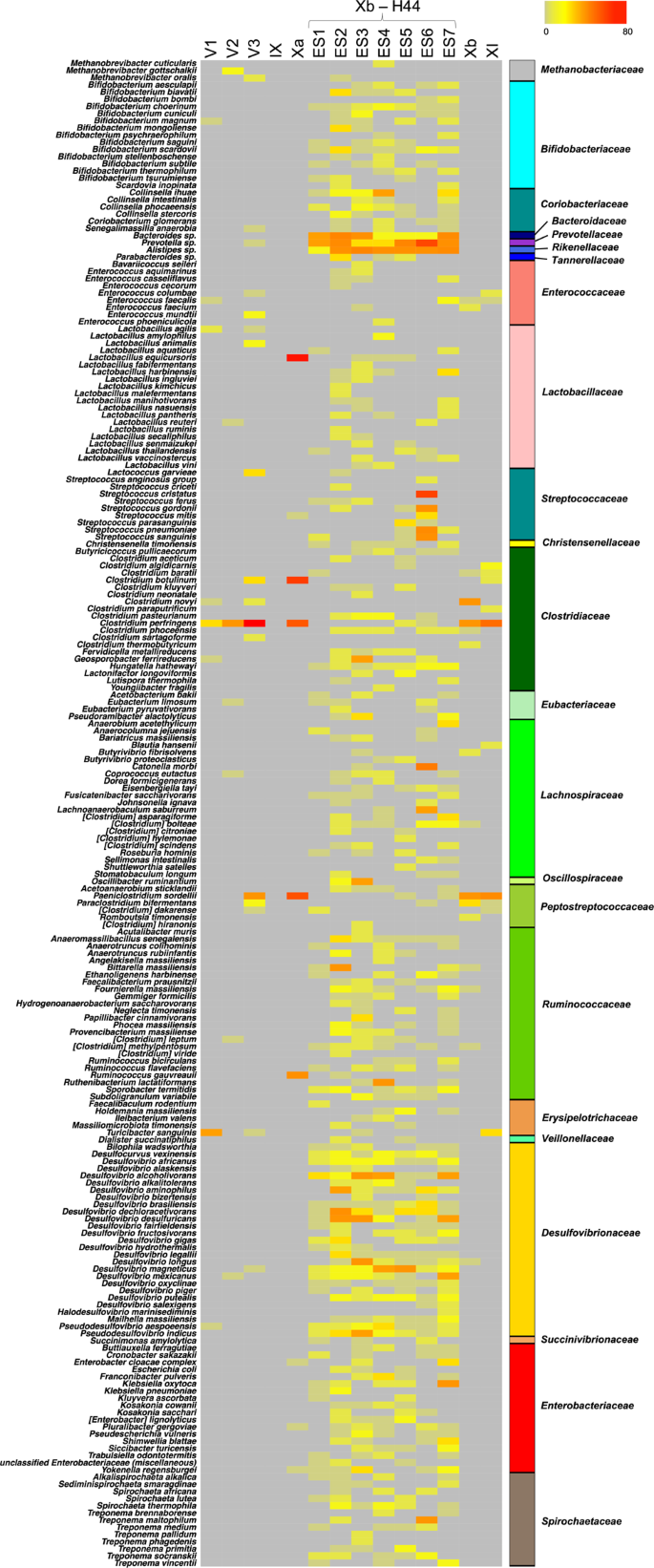 Further supporting a human-host origin of the bacterial species belonging to the hominid-associated gut microbiome families detected in the El Salt samples from SU IX, X and XI, feces or gastrointestinal tract are the first documented isolation source for 91 species out of 210 (43.3%), with 60 of these being classifiable as closely related to the human gut (Supplementary Data 3). In the latter subgroup, we can count several species from Lachnospiraceae (including well-known (beneficial) commensal inhabitants of modern human guts, such as Blautia, Coprococcus, Dorea, Fusicatenibacter and Roseburia spp.) and Ruminococcaeae families. Particularly, within Ruminococcaceae, we detected members of Anaerotruncus, Ruminococcus and Subdoligranulum genera, and the butyrate producer Faecalibacterium, one of the human commensal bacteria of greatest current interest, due to its very promising potential as a biomarker of a healthy gut microbiome48. Most of the aforementioned bacterial genera have been reported to account for the phylotypic diversity between human and non-human hominids, showing strong bias towards a human-host45. It is also worth remembering that most of these bacteria are able to produce short-chain fatty acids (mainly acetate and butyrate) from the fermentation of indigestible carbohydrates, through the establishment of complex syntrophic networks. Short-chain fatty acids are today considered metabolic and immunological gut microbiome players with a leading role in human physiology49. In addition, the Xb-H44 samples showed a high number of hits for Bacteroides, Parabacteroides, Alistipes and Bifidobacterium spp., other genera known to prevail in the human gut microbiome39,45. Interestingly, Bacteroides and Bifidobacterium have been shown to exhibit patterns of co-speciation with hominids45. For Bifidobacterium, this is particularly consistent with the propensity of this genus to be maternally inherited across generations. Being capable of metabolizing milk oligosaccharides and acting as a potent immunomodulator, the presence of vertically transmitted Bifidobacterium spp. in the infant gut could have provided important growth benefits to infant Hominidae50,51,52. To further characterize the ancient microbial taxa detected in the El Salt samples, we applied the HOPS53-based approach recently used by Jensen et al.30. In short, all the reads were first annotated and, subsequently, the ancient origin of each taxon was authenticated by computing three indicators: (i) the fraction of reads with PMDS > 1, (ii) the negative difference proportion (−Δ %) of PMDS > 1 reads, and (iii) their deamination rate at 5′. Taxa showing more than 200 assigned reads, more than 50 reads with PMDS > 1, −Δ % = 1 and C-T transition at 5′ >10% were considered to be of ancient origin (see Table 1 and Supplementary Figs. 6–8 for MapDamage plots, coverage plots and edit distance distribution)30. This in-depth characterization of the microbial metagenomic reads from the El Salt samples allowed us to confirm the presence of several species belonging to the gut microbiome families of hominids (including, among others, Alistipes, Bifidobacterium, Desulfovibrio and Prevotella spp., and Faecalibacterium prausnitzii), showing a read profile consistent with their ancient origin. As mentioned above, high amounts of coprostanol, a metabolite formed through hydrogenation of cholesterol by specific bacteria in the intestine of higher mammals, were found in some of the El Salt sediments from SU X, with proportions consistent with the presence of human fecal matter25. We therefore specifically looked for microorganisms capable of this metabolism in the aDNA from El Salt samples. To date, cholesterol-reducing capabilities associated with coprostanol conversion in feces have been suggested for Bifidobacterium, Collinsella, Bacteroides, Prevotella, Alistipes, Parabacteroides, Enterococcus, Lactobacillus, Streptococcus, Eubacterium, Lachnospiraceae (e.g., Coprococcus and Roseburia) and Ruminococcaceae (e.g., Anaerotruncus, Faecalibacterium, Ruminococcus and Subdoligranulum)54,55,56,57,58, which were all detected, at variable but substantial abundances, within the species belonging to the 24 gut microbiome families (as defined above) in Xa and Xb-H44 subunits. While lending support to the presence of coprostanol in the same layer as reported by Sistiaga et al.25, our findings on the representation of potential cholesterol-reducing bacteria in Neanderthal feces point to the microbial metabolism of cholesterol as an important function of the human gut microbiome for both modern and ancient humans, and suggest that relatively higher cholesterol intake has been a feature of the human diet at least since the Middle Pleistocene. Finally, the remaining bacterial species belonging to the hominid gut microbiome families identified in El Salt sediments from SUs IX, X and XI could be sorted into two major source categories: human (or animal) oral and/or pathobiont, and environmental (see Supplementary Fig. 9). In particular, possibly consistent with evidence of dental caries and periodontal disease in Neanderthals18, we found traces of potential opportunistic pathogens (e.g., Methanobrevibacter oralis, Scardovia inopinata, Streptococcus parasanguinis, Streptococcus sanguinis, Pseudoramibacter alactolyticus, Catonella morbi, Johnsonella ignava, Lachnoanaerobaculum saburreum, Shuttleworthia satelles, Stomatobaculum longum, Treponema maltophilum, Treponema medium, Treponema socranskii and Treponema vincentii), which have been associated with modern oral and dental diseases in humans59,60,61,62,63,64,65,66,67,68. Expectedly, the samples from the upper part of SU V (which are poor in archaeological remains) showed scarce and inconsistent presence of aDNA related to hominid-associated gut microbiome bacterial families. The highest hit counts were found for Clostridium perfringens, Paeniclostridium sordellii and Turicibacter sanguinis, with the first two being environmental opportunistic microorganisms historically associated with human gangrene and the last with acute appendicitis69,70,71. These findings further support the presence of potential human-like gut microbiome components as being unique to the samples from Xa and Xb, the only sedimentary layers that to date have shown traces of microscopic coprolites and fecal lipid biomarkers of presumed archaic human origin. In conclusion, by reconstructing ancient bacterial profiles from El Salt Neanderthal feces-containing sediments, we propose the existence of a core human gut microbiome with recognizable coherence between Neanderthals and modern humans, whose existence would pre-date the split between these two lineages, i.e., in the early Middle Pleistocene72. Although the risk of fractional contamination by modern DNA can never be ruled out and our data must be taken with some caution, the identification of this ancient human gut microbiome core supports the existence of evolutionary symbioses with strong potential to have a major impact on our health. In particular, the presence of known short-chain fatty acid producers, such as Blautia, Dorea, Roseburia, Rumunicoccus, Subdoligranulum, Faecalibacterium and Bifidobacterium, among the gut microbiome of Neanderthals, provides a unique perspective on their relevance as keystone taxa to the biology and health of the Homo lineage. While the former are known to allow extra energy to be extracted from dietary fiber73, strengthening the relevance of plant foods in human evolution, Bifidobacterium could have provided benefits to archaic human mothers and infants as a protective and immunomodulatory microorganism. Furthermore, the detection of so-called “old friend” microorganisms74 as putative components of Neanderthal gut microbiome (e.g., Spirochaetaceae, Prevotella and Desulfovibrio) further supports the hypothesized ancestral nature of these human gut microbiome members, which are now disappearing in westernized populations3,4,5,6,7,8,9,10,11. In the current scenario where we are witnessing a wholescale loss of bacterial diversity in the gut microbiome of the cultural “west”, with the parallel rise in dysbiosis-related autoimmune and inflammatory disorders75, the identification of evolutionarily integral taxa of the human holobiont may benefit practical applications favoring their retention among populations living in or transitioning to increasingly microbially deplete contexts. Such therapeutic applications may in the near future include next-generation probiotics, prebiotics or other gut microbiome-tailored dietary interventions. |
|
|
|
Post by Admin on Mar 3, 2021 23:32:45 GMT
The linguistic capacities in Neanderthals have long been an area of active research and debate among scientists, albeit with little resolution.
The last two decades have seen increasing archaeological discoveries documenting complex behaviors in this sister species to Homo sapiens. These have been linked to the possible presence of language, since it seems reasonable to suggest that such behaviors require the presence of a complex and efficient oral communication system.
Nevertheless, a different point of view maintains that the distinctive features of human language, absent in other organisms, include a symbolic element as well as a recursive syntactic process called merge.
This latter process, at its simplest, uses two syntactic elements and assembles them to form a set and is argued to be exclusive to Homo sapiens and to have appeared no earlier than 100,000 years ago.
Tracing the presence of symbolism and syntactic processes in the course of human evolution currently lies outside the realm of possibility in paleontology.
Nevertheless, the study of human fossils can prove key to determining whether past human species, and in particular the Neanderthals, possessed the anatomy necessary to produce and perceive an oral communication system as complex and efficient as human speech, the usual vehicle for language.
In other words, although paleontology cannot study the evolution of the ‘software’ of language it can contribute to our understanding of the evolution of the ‘hardware’ of speech.
“For decades, one of the central questions in human evolutionary studies has been whether the human form of communication, spoken language, was also present in any other species of human ancestor, especially the Neanderthals,” said Professor Juan Luis Arsuaga, a researcher at the Centro Mixto (UCM-ISCIII) de Evolución y Comportamiento Humanos and the Departamento de Geodinámica at the Universidad Complutense de Madrid.
Using high-resolution CT scans, Professor Arsuaga and his colleagues created 3D models of the ear structures of Homo sapiens, Neanderthals, and the Sima de los Huesos hominins, considered ancestors of the later Neanderthals.
They then entered the new data into a software-based model, developed in the field of auditory bioengineering, to estimate the hearing abilities up to 5 kHz, which encompasses most of the frequency range of modern human speech sounds.
Compared with the Sima de los Huesos hominins, Neanderthals showed slightly better hearing between 4-5 kHz, resembling modern humans more closely.
In addition, the researchers calculated the frequency range of maximum sensitivity, technically known as the occupied bandwidth, in each species.
“The occupied bandwidth is related to the communication system, such that a wider bandwidth allows for a larger number of easily distinguishable acoustic signals to be used in the oral communication of a species,” they explained.
“This, in turn, improves the efficiency of communication, the ability to deliver a clear message in the shortest amount of time.”
The Neanderthals had a wider bandwidth compared with their ancestors from Atapuerca, more closely resembling modern humans in this feature.
“This really is the key. The presence of similar hearing abilities, particularly the bandwidth, demonstrates that the Neanderthals possessed a communication system that was as complex and efficient as modern human speech,” said Professor Mercedes Conde-Valverde, a researcher in the Cátedra de Otoacústica Evolutiva y Paleoantropología at the Universidad de Alcalá.
“One of the other interesting results from the study was the suggestion that Neanderthal speech likely included an increased use of consonants,” said Professor Rolf Quam, a researcher in the Department of Anthropology at the Binghamton University, the Division of Anthropology at the American Museum of Natural History, the Universidad de Alcalá, and the Centro Mixto (UCM-ISCIII) de Evolución y Comportamiento Humanos.
The team’s results show that Neanderthals had a similar capacity to us to produce the sounds of human speech, and their ear was ‘tuned’ to perceive these frequencies.
This change in the auditory capacities in Neanderthals, compared with the ancestral Sima de los Huesos hominins, parallels archaeological evidence for increasingly complex behavioral patterns, including changes in stone tool technology, domestication of fire and possible symbolic practices.
“These results are particularly gratifying. We believe, after more than a century of research into this question, that we have provided a conclusive answer to the question of Neanderthal speech capacities,” said Dr. Ignacio Martinez, a researcher at the Centro Mixto (UCM-ISCIII) de Evolución y Comportamiento Humanos and the Departamento de Geodinámica at the Universidad Complutense de Madrid.
The findings were published in the journal Nature Ecology & Evolution.
Neanderthals and Homo sapiens had similar auditory and speech capacities
Abstract
The study of audition in fossil hominins is of great interest given its relationship with intraspecific vocal communication. While the auditory capacities have been studied in early hominins and in the Middle Pleistocene Sima de los Huesos hominins, less is known about the hearing abilities of the Neanderthals. Here, we provide a detailed approach to their auditory capacities. Relying on computerized tomography scans and a comprehensive model from the field of auditory bioengineering, we have established sound power transmission through the outer and middle ear and calculated the occupied bandwidth in Neanderthals. The occupied bandwidth is directly related to the efficiency of the vocal communication system of a species. Our results show that the occupied bandwidth of Neanderthals was greater than the Sima de los Huesos hominins and similar to extant humans, implying that Neanderthals evolved the auditory capacities to support a vocal communication system as efficient as modern human speech.
All the technical data regarding the CT scans as well as the measurements of 3D reconstructions necessary to reproduce our work are offered within the manuscript and Supplementary Information. CT scans of fossil material from Krapina are available at the Nespos platform. CT scans of the fossil specimens La Chapelle-aux-Saints 1 and La Quina H5 are the property of Musée de l’Homme (France); that for Amud 1 is the property of Tel Aviv University (Israel); and the fossil specimens from the Sima de los Huesos are property of Junta de Castilla y León (Spain), to whom application must be made for access. Interested readers may contact the authors, who will assist in getting in touch with the relevant institutions. The CT scans and 3D models of recent H. sapiens are available at Morphosource (https://www.morphosource.org/projects/000343670?locale=en).
|
|

