|
|
Post by Admin on Apr 9, 2021 3:03:23 GMT
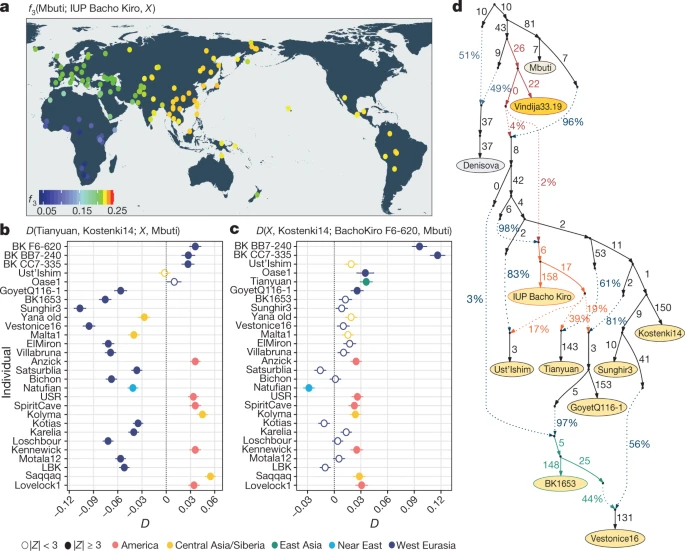 Fig. 2: Population affinities of the IUP Bacho Kiro Cave individuals. a, Allele sharing (f3) between the IUP Bacho Kiro Cave individuals and present-day populations (X) from the Simons Genome Diversity Project (SGDP)31 after their separation from an outgroup (Mbuti) (calculated as f3(Mbuti; IUP Bacho Kiro, X). Warmer colours on the map48 correspond to higher f3 values (higher shared genetic drift). b, IUP Bacho Kiro Cave individuals share significantly more alleles (proportions of allele sharing or D values plotted on x axis) with the roughly 40,000-year-old Tianyuan individual13 than with the approximately 38,000-year-old Kostenki14 individual29,30. Calculated as D(Tianyuan, Kostenki14; X, Mbuti). c, F6-620 shares significantly more alleles with the Oase17 and GoyetQ116-129 individuals, ancient Siberians and Native American individuals than with the Kostenki14 individual. Calculated as D(X, Kostenki14; F6-620, Mbuti). b, c, Filled circles indicate a significant value (|Z| ≥ 3); open circles, |Z| < 3. Whiskers correspond to 1 s.e. calculated across all autosomes (1,813,821 SNPs) using a weighted block jackknife28 and a block size of 5 Mb. BK, Bacho Kiro. d, Admixture graph relating Bacho Kiro Cave individuals and ancient humans older than 30 kyr BP. This model uses 281,732 overlapping SNPs in all individuals and fits the data with a single outlier (Z = 3.22). Ancient non-Africans (yellow circles), Vindija 33.19 Neanderthal (orange), Denisovan (grey) and present-day African individuals (light yellow circle) are shown. Admixture edges (dotted lines) show the genetic component related to Neanderthals (red), to the IUP Bacho Kiro Cave individuals (orange) and to BK1653 (green). Numbers on solid branches correspond to the estimated drift in f2 units of squared frequency difference; labels on dotted edges give admixture proportions. We next investigated whether these observations could be due to the fact that present-day populations in western Eurasia derive part of their ancestry from ‘Basal Eurasians’32,33, an inferred population that diverged early from other non-African populations and may have ‘diluted’ allele sharing between western Eurasian populations and IUP individuals. To do this, we compared the Ust’Ishim, Oase1 and IUP Bacho Kiro Cave individuals to western Eurasian individuals such as the approximately 38,000-year-old ‘Kostenki14’ individual from Russia29,30, which pre-dates the introduction of ‘Basal Eurasian’ ancestry to Europe around 8,000 cal. BP32. We found that the Ust’Ishim and Oase1 individuals showed no more affinity to western than to eastern Eurasian populations, suggesting that they did not contribute ancestry to later Eurasian populations, as previously shown7,8 (Supplementary Information 5, Extended Data Fig. 5). By contrast, the IUP Bacho Kiro Cave individuals shared more alleles with the roughly 40,000-year-old Tianyuan individual13 from China (Fig. 2b) and other ancient Siberians34,35 and Native Americans36,37,38,39 (Fig. 2c) than with the Kostenki14 individual (3.6 ≤ |Z| ≤ 5.3). Among other western Eurasian Upper Palaeolithic humans, the IUP Bacho Kiro Cave individuals shared more alleles with the Oase1 (3.6 ≤ |Z| ≤ 4.3) and roughly 35,000-year-old GoyetQ116-129 individuals than with the Kostenki14 individual (3.2 ≤ |Z| ≤ 4.3; Fig. 2c, Supplementary Information 5). Notably, the GoyetQ116-1 individual has previously been shown to share more alleles with early East Asians than other individuals of a similar age in Europe13. When we explored models of population history that are compatible with the observations above using admixture graphs28, we found that the IUP Bacho Kiro Cave individuals were related to populations that contributed ancestry to the Tianyuan individual in China as well as, to a lesser extent, to the GoyetQ116-1 and Ust’Ishim individuals (all |Z| < 3; Fig. 2d, Supplementary Information 6). This resolves the previously unclear relationship between the GoyetQ116-1 and Tianyuan individuals13 without the need for gene flow between these two geographically distant individuals. The models also suggest that the later BK1653 individual belonged to a population that was related, but not identical, to that of the GoyetQ116-1 individual (Fig. 2d, Extended Data Fig. 4, Supplementary Information 6) and that the Věstonice cluster, whose members were found in association with Gravettian assemblages29, derived part of their ancestry from such a population and the rest from populations related to the roughly 34,000-year-old ‘Sunghir’ individuals40 from Russia (Fig. 2d, Supplementary Information 6). As the IUP Bacho Kiro Cave individuals lived at the same time as some of the last Neanderthals in Europe6, we estimated the proportion of Neanderthal DNA in their genomes by taking advantage of two high-quality Neanderthal genomes9,10,41. We found that the IUP individuals F6-620, BB7-240 and CC7-335 carried 3.8% (95% confidence interval (CI): 3.3–4.4%), 3.0% (95% CI: 2.4–3.6%) and 3.4% (95% CI: 2.8–4.0%) Neanderthal DNA, respectively. This is more than the average of 1.9% (95% CI: 1.5–2.4%) found in other ancient or present-day humans, except for the Oase1 individual, who had a close Neanderthal relative (6.4% (95% CI: 5.7–7.1%); Extended Data Fig. 6, Supplementary Information 7). By contrast, the more recent BK1653 individual carried only 1.9% (95% CI: 1.4–2.4%) Neanderthal DNA, similar to other ancient and present-day humans10,41 (Extended Data Fig. 6). As has been the case for all humans studied so far, the Neanderthal DNA in BK1653 and the IUP Bacho Kiro Cave individuals was more similar to the Vindija33.1910 and Chagyrskaya842 Neanderthals than to the Altai Neanderthal9 (2.8 ≤ |Z| ≤ 5.1; Supplementary Information 7). |
|
|
|
Post by Admin on Apr 9, 2021 20:23:57 GMT
To study the spatial distribution of Neanderthal ancestry in the genomes of the Bacho Kiro Cave individuals, we used around 1.7 million SNPs at which Neanderthal9 and/or Denisovan43 genomes differ from African genomes7 and an approach44 that detects tracts of archaic DNA in ancient genomes. We found a total of 279.6 centiMorgans (cM) of Neanderthal DNA in F6-620, 251.6 cM in CC7-335 and 220.9 cM in BB7-240, and these individuals carried seven, six and nine Neanderthal DNA segments longer than 5 cM, respectively (Fig. 3, Extended Data Fig. 7a, Supplementary Information 8). The longest introgressed Neanderthal segment in F6-620 encompassed 54.3 cM, and the longest segments in CC7-335 and BB7-240 were 25.6 cM and 17.4 cM, respectively (Fig. 3, Extended Data Fig. 7a). By contrast, the total amount of Neanderthal DNA in the BK1653 genome was 121.7 cM and the longest Neanderthal segment was 2.5 cM (Fig. 3, Extended Data Fig. 7a). Fig. 3: Geographical distribution of Neanderthal archaeological sites and genome-wide distribution of Neanderthal alleles in the genomes of ancient modern humans. 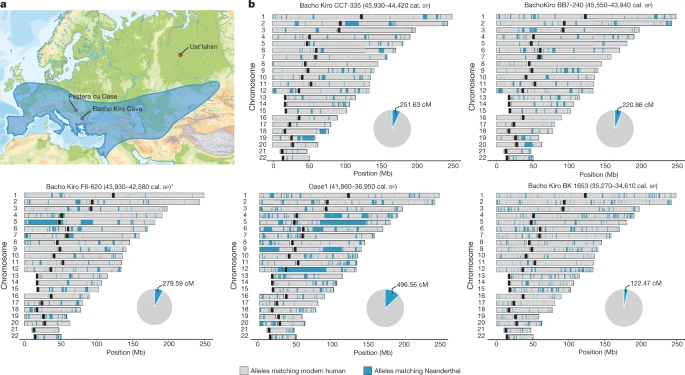 a, Neanderthal geographical range (blue) and the locations of Peştera cu Oase, Bacho Kiro Cave and where the femur of the Ust’Ishim individual was found. b, Distribution of Neanderthal DNA in ancient modern human genomes. Neanderthal DNA segments longer than 0.2 cM are indicated in blue. Pie charts indicate the total proportion of Neanderthal DNA identified in each genome. Centromeres are shown in black. On the basis of the distribution of the long Neanderthal segments, we estimate that individual F6-620 had a Neanderthal ancestor less than six generations back in his family tree (Extended Data Table 2, Supplementary Information 8). Both the CC7-335 and BB7-240 individuals had Neanderthal ancestors about seven generations back in their families, with upper confidence intervals of ten and seventeen generations, respectively (Extended Data Table 2, Extended Data Fig. 7b, Supplementary Information 8). Thus, all IUP Bacho Kiro Cave individuals had recent Neanderthal ancestors in their immediate family histories. To further explore the extent to which the Bacho Kiro Cave individuals contributed ancestry to later populations in Eurasia, we investigated whether the Neanderthal DNA segments in Bacho Kiro Cave genomes overlapped with Neanderthal segments in present-day populations more than expected by chance. We found more overlapping of segments between present-day East Asian populations and the IUP Bacho Kiro Cave individuals (lowest correlation coefficient of 0.09, 95% CI: 0.08–0.1) than with the BK1653 individual (P = 0.02, Wilcoxon test). By contrast, the BK1653 individual shows more overlapping of Neanderthal segments with present-day western Eurasian populations (a correlation coefficient of 0.11, 95% CI: 0.1–0.12) than do the IUP Bacho Kiro Cave individuals (P < 1 × 10−18, Wilcoxon test). This is compatible with the observation that the IUP Bacho Kiro Cave population contributed more ancestry to later populations in Asia and the Americas, whereas the BK1653 individual contributed more ancestry to populations in western Eurasia. We next looked for overlap between parts of the human genome that carry little or no Neanderthal ancestry (Neanderthal ‘deserts’), which are thought to have been caused by purifying selection against Neanderthal DNA shortly after introgression45,46. We find almost no introgressed Neanderthal DNA in the previously described deserts in the IUP Bacho Kiro Cave and Oase1 individuals (249 kb out of 898 Mb of introgressed sequence; P = 0.0079, permutation P value). When we restricted these comparisons to the more recent Neanderthal contributions (that is, segments longer than 5 cM), we similarly found no overlap (0 Mb out of 415 Mb, P = 0.15, permutation P value), suggesting that selection against Neanderthal DNA variants occurred within a few generations, although additional individuals with recent Neanderthal ancestry will be needed to fully resolve this question. In conclusion, the Bacho Kiro Cave genomes show that several distinct modern human populations existed during the early Upper Palaeolithic in Eurasia. Some of these populations, represented by the Oase1 and Ust’Ishim individuals, show no detectable affinities to later populations, whereas groups related to the IUP Bacho Kiro Cave individuals contributed to later populations with Asian ancestry as well as some western Eurasian humans such as the GoyetQ116-1 individual in Belgium. This is consistent with the fact that IUP archaeological assemblages are found from central and eastern Europe to present-day Mongolia5,15,16 (Fig. 1), and a putative IUP dispersal that reached from eastern Europe to East Asia. Eventually populations related to the IUP Bacho Kiro Cave individuals disappeared in western Eurasia without leaving a detectable genetic contribution to later populations, as indicated by the fact that later individuals, including BK1653 at Bacho Kiro Cave, were closer to present-day European populations than to present-day Asian populations29,30. In Europe, the notion of successive population replacements is also consistent with the archaeological record, where the IUP is clearly intrusive against the Middle Palaeolithic background and where, apart from the common focus on blades, there are no clear technological connections between the IUP and the subsequent Aurignacian technologies. Finally, it is striking that all four of the European individuals who overlapped in time with late Neanderthals7 and from whom genome-wide data have been retrieved had close Neanderthal relatives in their family histories (Fig. 3, Extended Data Figs. 7, 8). This suggests that mixing between Neanderthals and the first modern humans that arrived into Europe was perhaps more common than is often assumed. |
|
|
|
Post by Admin on Apr 19, 2021 2:40:46 GMT
Skeletal remains are important sources of Pleistocene hominin DNA, but are rarely recovered at archaeological sites. Mitochondrial DNA (mtDNA) has been retrieved from cave sediments, but provides limited value for studying population relationships.  Although nuclear DNA contains far more information, its retrieval from sediments presents substantial challenges. It’s far less abundant than mtDNA and difficult to distinguish from other non-hominin mammalian and microbial DNA, which dominates the genetic material often present in ancient sediments. To address these challenges, Dr. Benjamin Vernot from the Department of Evolutionary Genetics at the Max-Planck-Institute for Evolutionary Anthropology and his colleagues developed methods to recover, enrich and analyze nuclear DNA from cave sediments.  “There are lots of places in the human genome that are very similar to a bear’s DNA, for example,” Dr. Vernot said. “We specifically targeted regions in the genome where we could be confident of isolating only human DNA, and we also designed methods to measure our success in removing non-human DNA.”  “We wanted to be confident that we weren’t accidentally looking at some unknown species of hyena.” Specifically, the researchers applied their approach to extract nuclear DNA from more than 150 sediment samples from three caves: Galería de las Estatuas in northern Spain and Chagyrskaya and Denisova caves in the Altai Mountains of southern Siberia. They detected a population replacement in Spain approximately 100,000 years ago, accompanied by a turnover of mtDNA. They also identified two radiation events in Neanderthal history during the early part of the Late Pleistocene. “The dawn of nuclear DNA analysis of sediments massively extends the range of options to tease out the evolutionary history of ancient humans,” Dr. Vernot said.  “By freeing the field of ancient DNA from the constraints of finding human remains and expanding the number of sites potentially suitable for investigation, we can now study the DNA from many more human populations, and from many more places, than has previously been thought possible,” added Dr. Matthias Meyer, also from the Department of Evolutionary Genetics at the Max-Planck-Institute for Evolutionary Anthropology. The findings were published in the journal Science. Abstract Bones and teeth are important sources of Pleistocene hominin DNA, but are rarely recovered at archaeological sites. Mitochondrial DNA has been retrieved from cave sediments, but provides limited value for studying population relationships. We therefore developed methods for the enrichment and analysis of nuclear DNA from sediments, and applied them to cave deposits in western Europe and southern Siberia dated to between approximately 200,000 and 50,000 years ago. We detect a population replacement in northern Spain approximately 100,000 years ago, accompanied by a turnover of mitochondrial DNA. We also identify two radiation events in Neanderthal history during the early part of the Late Pleistocene. Our work lays the ground for studying the population history of ancient hominins from trace amounts of nuclear DNA in sediments. _____ Benjamin Vernot et al. Unearthing Neanderthal population history using nuclear and mitochondrial DNA from cave sediments. Science, published online April 15, 2021; doi; 10.1126/science.abf1667 |
|
|
|
Post by Admin on May 12, 2021 20:05:41 GMT
A new study looking at the evolutionary history of the human oral microbiome shows that Neanderthals and ancient humans adapted to eating starch-rich foods as far back as 100,000 years ago, which is much earlier than previously thought.
The findings suggest such foods became important in the human diet well before the introduction of farming and even before the evolution of modern humans. And while these early humans probably didn't realize it, the benefits of bringing the foods into their diet likely helped pave the way for the expansion of the human brain because of the glucose in starch, which is the brain's main fuel source.
"We think we're seeing evidence of a really ancient behavior that might have been part encephalization -- or the growth of the human brain," said Harvard Professor Christina Warinner, Ph.D. '10. "It's evidence of a new food source that early humans were able to tap into in the form of roots, starchy vegetables, and seeds."
The findings come from a seven-year study published in the Proceedings of the National Academy of Sciences on Monday that involved the collaboration of more than 50 international scientists. Researchers reconstructed the oral microbiomes of Neanderthals, primates, and humans, including what's believed to be the oldest oral microbiome ever sequenced -- a 100,000-year-old Neanderthal.
The goal was to better understand how the oral microbiome -- a community of microorganisms in our mouths that help to protect against disease and promote health -- developed since little is known about its evolutionary history.
"For a long time, people have been trying to understand what a normal healthy microbiome is," said Warinner, assistant professor of anthropology in the Faculty of Arts and Sciences and the Sally Starling Seaver Assistant Professor at the Radcliffe Institute. "If we only have people today that we're analyzing from completely industrialized contexts and that already have high disease burdens, is that healthy and normal? We started to ask: What are the core members of the microbiome? Which species and groups of bacteria have actually co-evolved with us the longest?"
The scientists analyzed the fossilized dental plaque of both modern humans and Neanderthals and compared them to those of humanity's closest primate relatives, chimpanzees and gorillas, as well as howler monkeys, a more distant relative.
Using newly developed tools and methods, they genetically analyzed billions of DNA fragments preserved in the fossilized plaque to reconstruct their genomes. It's similar in theory to how archeologists painstakingly piece together ancient broken pots, but on a much larger scale.
The biggest surprise from the study was the presence of particular strains of oral bacteria that are specially adapted to break down starch. These strains, which are members of the genus Streptococcus, have a unique ability to capture starch-digesting enzymes from human saliva, which they then use to feed themselves. The genetic machinery the bacteria uses to do this is only active when starch is part of the regular diet.
Both the Neanderthals and the ancient humans scientists studied had these starch-adapted strains in their dental plaque while most of the primates had almost no streptococci that could break down starch.
"It seems to be a very human specific evolutionary trait that our Streptococcus acquired the ability to do this," Warinner said.
The findings also push back on the idea that Neanderthals were top carnivores, given that the "brain requires glucose as a nutrient source and meat alone is not a sufficient source," Warinner said.
Researchers said the finding makes sense because for hunter-gatherer societies around the world, starch-rich foods -- underground roots, tubers (like potatoes), and forbs, as well as nuts and seeds, for example -- are important and reliable nutrition sources. In fact, starch currently makes up about 60 percent of calories for humans worldwide.
"Its availability is much more predictable across the annual season for tropical hunter-gatherers," said Richard W. Wrangham, Ruth B. Moore Professor of Biological Anthropology and one of the paper's co-authors. "These new data make every sense to me, reinforcing the newer view about Neanderthals that their diets were more sapien-like than once thought, [meaning] starch-rich and cooked."
The research also identified 10 groups of bacteria that have been part of the human and primate oral microbiome for more than 40 million years and are still shared today. While these bacteria may serve important and beneficial roles, relatively little is known about them. Some don't even have names.
Focusing on Neanderthals and today's humans, the analysis surprisingly showed the oral microbiome of both groups were almost indistinguishable. Only when looking at individual bacterial strains could they see some differences. For example, ancient humans living in Europe before 14,000 years ago during the Ice Age shared some bacterial strains with Neanderthals that are no longer found in humans today.
The differences and similarities from the study are all part of what makes us human, Warinner said. It also touches on the power of analyzing the tiny microbes that live in the human body, she said.
"It shows that our microbiome encodes valuable information about our own evolution that sometimes gives us hints at things that otherwise leave no traces at all," Warinner said.
Story Source:
Materials provided by Harvard University. Original written by Juan Siliezar. Note: Content may be edited for style and length.
Journal Reference:
James A. Fellows Yates, Irina M. Velsko, Franziska Aron, Cosimo Posth, Courtney A. Hofman, Rita M. Austin, Cody E. Parker, Allison E. Mann, Kathrin Nägele, Kathryn Weedman Arthur, John W. Arthur, Catherine C. Bauer, Isabelle Crevecoeur, Christophe Cupillard, Matthew C. Curtis, Love Dalén, Marta Díaz-Zorita Bonilla, J. Carlos Díez Fernández-Lomana, Dorothée G. Drucker, Elena Escribano Escrivá, Michael Francken, Victoria E. Gibbon, Manuel R. González Morales, Ana Grande Mateu, Katerina Harvati, Amanda G. Henry, Louise Humphrey, Mario Menéndez, Dušan Mihailović, Marco Peresani, Sofía Rodríguez Moroder, Mirjana Roksandic, Hélène Rougier, Sandra Sázelová, Jay T. Stock, Lawrence Guy Straus, Jiří Svoboda, Barbara Teßmann, Michael J. Walker, Robert C. Power, Cecil M. Lewis, Krithivasan Sankaranarayanan, Katerina Guschanski, Richard W. Wrangham, Floyd E. Dewhirst, Domingo C. Salazar-García, Johannes Krause, Alexander Herbig, Christina Warinner. The evolution and changing ecology of the African hominid oral microbiome. Proceedings of the National Academy of Sciences, 2021; 118 (20): e2021655118 DOI: 10.1073/pnas.2021655118
|
|
|
|
Post by Admin on May 13, 2021 3:20:03 GMT
The evolution and changing ecology of the African hominid oral microbiome PNAS May 18, 2021 118 (20) e2021655118; doi.org/10.1073/pnas.2021655118Edited by Robert R. Dunn, North Carolina State University, Raleigh, NC, and accepted by Editorial Board Member James F. O’Connell March 22, 2021 (received for review October 16, 2020) Significance The microbiome plays key roles in human health, but little is known about its evolution. We investigate the evolutionary history of the African hominid oral microbiome by analyzing dental biofilms of humans and Neanderthals spanning the past 100,000 years and comparing them with those of chimpanzees, gorillas, and howler monkeys. We identify 10 core bacterial genera that have been maintained within the human lineage and play key biofilm structural roles. However, many remain understudied and unnamed. We find major taxonomic and functional differences between the oral microbiomes of Homo and chimpanzees but a high degree of similarity between Neanderthals and modern humans, including an apparent Homo-specific acquisition of starch digestion capability in oral streptococci, suggesting microbial coadaptation with host diet. Abstract The oral microbiome plays key roles in human biology, health, and disease, but little is known about the global diversity, variation, or evolution of this microbial community. To better understand the evolution and changing ecology of the human oral microbiome, we analyzed 124 dental biofilm metagenomes from humans, including Neanderthals and Late Pleistocene to present-day modern humans, chimpanzees, and gorillas, as well as New World howler monkeys for comparison. We find that a core microbiome of primarily biofilm structural taxa has been maintained throughout African hominid evolution, and these microbial groups are also shared with howler monkeys, suggesting that they have been important oral members since before the catarrhine–platyrrhine split ca. 40 Mya. However, community structure and individual microbial phylogenies do not closely reflect host relationships, and the dental biofilms of Homo and chimpanzees are distinguished by major taxonomic and functional differences. Reconstructing oral metagenomes from up to 100 thousand years ago, we show that the microbial profiles of both Neanderthals and modern humans are highly similar, sharing functional adaptations in nutrient metabolism. These include an apparent Homo-specific acquisition of salivary amylase-binding capability by oral streptococci, suggesting microbial coadaptation with host diet. We additionally find evidence of shared genetic diversity in the oral bacteria of Neanderthal and Upper Paleolithic modern humans that is not observed in later modern human populations. Differences in the oral microbiomes of African hominids provide insights into human evolution, the ancestral state of the human microbiome, and a temporal framework for understanding microbial health and disease. The oral cavity is colonized by one of the most diverse sets of microbial communities of the human body, currently estimated at over 600 prevalent taxa (1). Dental diseases, such as caries and periodontitis, remain health burdens in all human populations despite hygiene interventions (2, 3), and oral microbes are often implicated in extraoral inflammatory diseases (4, 5). To date, most oral microbiome research has focused on clinical samples obtained from industrialized populations that have daily oral hygiene routines and access to antibiotics (1, 6), but far less is known about the global diversity of the oral microbiome, especially from diverse past and present nonindustrialized societies (7). The oral cavity contains at least six distinct habitats, but dental biofilms, including both supra- and subgingival dental plaque, are among the most diverse and clinically important (1, 6, 8). During life, these dental biofilms naturally and repeatedly calcify, forming dental calculus (tooth tartar) (9), a robust, long-term record of the oral microbiome (10). Archaeological dental calculus has been shown to preserve authentic oral bacterial metagenomes in a wide range of historic and prehistoric populations and up to 50 thousand years ago (ka) (10⇓⇓–13). As such, dental calculus presents an opportunity to directly investigate the evolution of the hominid microbiome and to reconstruct ancestral states of the modern human oral microbiome. In addition, because research has shown that evolutionary traits, diet, and cultural behaviors shape modern human microbiome structure and function at other body sites, such as the gut and skin microbiomes (14⇓⇓⇓–18), investigating ancient oral metagenomes has the potential to reveal valuable information about major events in modern human evolution and prehistory, such as predicted dietary changes during the speciation of Homo (19⇓–21) and the direct interaction of Neanderthals and modern humans during the Late Pleistocene (22). To better understand the evolutionary ecology of the African hominid microbiome, we generated and analyzed 109 dental calculus metagenomes from present-day modern humans (n = 8), gorillas (Gorilla, n = 29), chimpanzees (Pan, n = 20), Neanderthals (n = 13), and two groups of archaeological modern humans associated with major lifestyle transitions (preagricultural, n = 20; preantibiotic, n = 14), as well as New World howler monkeys (n = 5) for comparison (SI Appendix, Fig. S1). To account for potential sampling biases, we analyzed multiple subspecies and populations of each African great ape genus, which were obtained from C20th or C21st-collected museum collections, and for modern humans we sampled multiple populations from both Africa and Europe. To this, we added previously published microbiome data from chimpanzees (n = 1) (13), Neanderthals (n = 4) (13), and present-day modern humans (n = 10) (23), for a total dataset of 124 individuals (Fig. 1A, SI Appendix, Table S1, and Dataset S1). We also generated eight new radiocarbon dates for archaeological individuals, for a total of 44 directly or indirectly dated ancient individuals in this study (Dataset S1).  Fig. 1. Sample locations and oral microbiome authentication of ancient dental calculus. (A) Sample locations. (B) PCoA comparing euclidean distances of microbial genera of well-preserved ancient and present-day dental calculus to environmental proxy controls (degraded archaeological bone) and present-day dental plaque and feces. Ancient dental calculus is distinct from gut and archaeological bone but overlaps with present-day dental plaque. (C) Representative DNA damage patterns for Neanderthals and ancient and present-day modern humans for four oral-specific bacterial species. The Neanderthal and upper Paleolithic modern human individuals show expected damage patterns consistent with authentic aDNA, whereas the present-day individual does not. See also SI Appendix, Fig. S4. Here, we investigate the structure, function, and core microbial members of the human oral microbiome within an evolutionary framework, seeking to determine whether a core microbiome can be defined for each African hominid group, whether the core is phylogenetically coherent, and whether some members of the core are specific to certain host groups. We test whether the oral microbiome of hominids reflects host phylogeny, finding that African hominid oral microbiota are distinguished by major taxonomic and functional differences that only weakly reflect host relationships and are likely influenced by other physiological, dietary, or behavioral factors. We compare the microbial profiles of Neanderthals and modern humans and, contrary to expectations (12, 13), find a high consistency of oral microbiome structure within Homo, regardless of geography, time period, or diet/lifestyle. We detect the persistence of shared genetic diversity in core taxa between Neanderthals and Upper Paleolithic humans prior to 14 ka, supporting a growing body of evidence for earlier admixture and interaction in Ice Age Europe (24, 25). Finally, we explore possible implications of our findings on Homo-associated encephalization (19, 26) and the role of dietary starch in human evolution (20, 21) by investigating the evolutionary history of amylase-binding capability by oral streptococci. We find that amylase binding is an apparent Homo-specific trait, suggestive of microbial coadaptation to starch-rich diets early in human evolution. |
|

