|
|
Post by Admin on Feb 20, 2021 3:46:07 GMT
Conclusions.
A Neandertal haplotype on chromosome 12 is protective for severe disease in the current SARS-CoV-2 pandemic. It is present in populations in Eurasia and the Americas at carrier frequencies that often reach and exceed 50%. The ancestral Neandertal OAS locus variants may thus have been advantageous to modern humans throughout Eurasia, perhaps due to one or many epidemics involving RNA viruses, especially given that the Neandertal haplotype has been found to be protective for at least three RNA viruses (West Nile virus, hepatitis C virus, SARS-CoV). Supporting this notion, simulations have demonstrated that the Neandertal OAS haplotype has been under positive selection in modern humans (35). Strikingly, the OAS1 protein encoded by the modern human OAS haplotype is of lower enzymatic activity than the one encoded by the Neandertal haplotype (37). This may have been advantageous at some point in Africa, because loss-of-function mutations of the OAS1 locus have occurred numerous times among primates (45), suggesting that the maintenance of OAS1 activity is costly to an organism. One may speculate that, when modern humans encountered new RNA viruses outside Africa, the higher enzymatic activity of the ancestral variants that they acquired through genetic interactions with Neandertals may have been advantageous.
Intriguingly, there is evidence that the Neandertal-like OAS haplotype may have recently increased in frequency in Eurasia (Fig. 4A), suggesting that selection may have positively affected the Neandertal-derived OAS locus in the last millennium. Future studies of human remains from historical times will clarify whether, and when, this occurred.
Materials and Methods
The index variants for the seven novel loci (rs9380142, rs143334143, rs3131294, rs10735079, rs74956615, rs2109069, and rs2236757) were obtained from GenOMICC (22). The regional summary statistics from the round 4 release of the metaanalysis carried out by the COVID-19 HGI (24) (https://covid19hg.org/results) was used to analyze the chromosome 12 locus (hospitalized vs. population controls, i.e., “B2” phenotype, using all ancestries but not including the 23andMe study, due to limited release of number of variants). LD was calculated using LDlink 4.1, and alleles were compared to the archaic genomes using tabix (HTSlib 1.10). The haplotype associated with protection against severe COVID-19 was investigated using phylogenetic software (PhyML 3.0), and the probability of observing a haplotype of a certain length or longer due to incomplete lineage sorting was calculated as described (29). The present-day haplotypes were constructed by including all variable positions in the region chr12: 113,350,796 to 113,425,679, excluding singletons. Haplotypes seen more than 10 times were included in the phylogenetic analysis. The inferred ancestral states at variable positions among present-day humans were taken from Ensembl. Genotypes of ancient genomes of modern humans were obtained from a compiled database (42). Maps displaying allele frequencies of different populations were made using Mathematica 11.0 (Wolfram Research, Inc.) and OpenStreetMap data.
|
|
|
|
Post by Admin on Mar 7, 2021 0:31:44 GMT
A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity
Abstract
To identify circulating proteins influencing Coronavirus Disease 2019 (COVID-19) susceptibility and severity, we undertook a two-sample Mendelian randomization (MR) study, rapidly scanning hundreds of circulating proteins while reducing bias due to reverse causation and confounding. In up to 14,134 cases and 1.2 million controls, we found that an s.d. increase in OAS1 levels was associated with reduced COVID-19 death or ventilation (odds ratio (OR) = 0.54, P = 7 × 10−8), hospitalization (OR = 0.61, P = 8 × 10−8) and susceptibility (OR = 0.78, P = 8 × 10−6). Measuring OAS1 levels in 504 individuals, we found that higher plasma OAS1 levels in a non-infectious state were associated with reduced COVID-19 susceptibility and severity. Further analyses suggested that a Neanderthal isoform of OAS1 in individuals of European ancestry affords this protection. Thus, evidence from MR and a case–control study support a protective role for OAS1 in COVID-19 adverse outcomes. Available pharmacological agents that increase OAS1 levels could be prioritized for drug development.
Main
To date, the COVID-19 pandemic has caused more than 2 million deaths worldwide and infected approximately 100 million individuals1. Despite the scale of the epidemic, there are, at present, few disease-specific therapies2 to reduce the morbidity and mortality of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Apart from dexamethasone therapy in oxygen-dependent patients3, most clinical trials have shown, at most, mild or inconsistent benefits on disease outcomes4,5,6. Therefore, validated targets are needed for COVID-19 therapeutic development.
One source of such targets is circulating proteins. Recent advances in large-scale proteomics have enabled the measurement of thousands of circulating proteins—and when combined with evidence from human genetics, such targets greatly improve the probability of drug development success7,8,9. Although de novo drug development will take time, the repurposing of currently available molecules targeting those proteins could provide an accelerated opportunity to deliver new therapies to patients.
Nevertheless, because confounding and reverse causation often bias traditional circulating protein studies, methods are needed to dissect causal relationships. This is especially the case in COVID-19, where exposure to SARS-CoV-2 unleashes profound changes in circulating protein levels10. One way to address these limitations is by using MR, a genetic epidemiology method that uses genetic variants as instrumental variables to test the effect of an exposure (here, protein levels) on an outcome (here, COVID-19 outcomes). The process of random assignment of alleles at conception greatly reduces bias from confounding. Because genotypes are always assigned before disease onset, MR studies are not influenced by reverse causation. However, MR rests on several assumptions11, the most problematic being horizontal pleiotropy of the genetic instruments (wherein the genotype influences the outcome, independently of the exposure). One way to help avoid this bias is to use genetic variants that influence circulating protein levels that are adjacent to the gene that encodes the circulating protein through the use of cis-protein quantitative trait loci (cis-pQTLs)9. cis-pQTLs are likely to influence the level of the circulating protein by directly influencing its transcription or translation and, therefore, less likely to affect the outcome of interest through pleiotropic pathways. Nevertheless, a causal genetic association between the exposure and outcome might be confounded by linkage disequilibrium (LD)12, which can be detected through co-localization testing.
Understanding the etiologic role of circulating proteins in infectious diseases is challenging because the infection itself often leads to large changes in circulating protein levels10. Thus, it might appear that an increase in a circulating protein, such as a cytokine, is associated with a worsened outcome, when, in fact, the cytokine might be the host’s response to this infection and help to mitigate this outcome. It is, therefore, important to identify genetic determinants of the protein levels in the non-infected state, which would reflect a person’s baseline predisposition to the level of a protein.
MR studies can be complemented by traditional case–control studies, where the protein is longitudinally measured in patients with COVID-19 and controls, allowing for an estimation of the association between the protein level and COVID-19 outcomes. However, MR studies tend to predict the effect of the protein in the non-infectious state when the genetic determinants of such proteins are measured in the non-infected population. Because MR and case–control studies rely on different assumptions and might be influenced by different biases, concordant results between the two study designs can strengthen the cumulative evidence13.
In this study, we, therefore, undertook two-sample MR and co-localization analyses to combine results from large-scale genome-wide association studies (GWASs) of circulating protein levels and COVID-19 outcomes14. We began by identifying the genetic determinants of circulating protein levels in large-scale proteomic GWASs and then used MR to assess whether these cis-pQTLs were associated with COVID-19 outcomes in large COVID-19 GWASs. Next, we investigated expression QTL (eQTL) and splice QTL (sQTL) effects of lead proteins. We then measured the most promising protein, OAS1, in individuals ascertained for SARS-CoV-2 infection, followed for longitudinal sampling during and after their infection.
|
|
|
|
Post by Admin on Mar 7, 2021 4:56:08 GMT
Results MR using cis-pQTLs and pleiotropy assessment The study design is illustrated in Fig. 1. We began by obtaining the genetic determinants of circulating protein levels from six large proteomic GWASs of individuals of European ancestry (Sun et al.15 n = 3,301; Emilsson et al.16 n = 3,200; Pietzner et al.17 n = 10,708; Folkersen et al.18 n = 3,394; Yao et al.19 n = 6,861 and Suhre et al.20 n = 997). A total of 931 proteins from these six studies had genome-wide significant cis-pQTLs or highly correlated LD proxies (r2 > 0.8) in the meta-analyses of data from the COVID-19 Host Genetics Initiative21, which included results from the GenOMICC program22. We then undertook MR analyses using 1,425 cis-pQTLs and 39 LD proxies as genetic instruments for circulating proteins in three COVID-19 outcomes: 1) very severe COVID-19 disease (defined as individuals experiencing death, mechanical ventilation, non-invasive ventilation, high-flow oxygen or use of extra-corporeal membrane oxygenation; 99.7% of these individuals were of European ancestry) using 4,336 cases and 623,902 controls; 2) COVID-19 disease requiring hospitalization using 6,406 cases and 902,088 controls of European ancestry; and 3) COVID-19 susceptibility using 14,134 cases and 1,284,876 controls of European ancestry. In all outcomes, cases required evidence of SARS-CoV-2 infection. For the very severe COVID-19 and hospitalization outcomes, COVID-19 cases were defined as laboratory-confirmed SARS-CoV-2 infection based on nucleic acid amplification or serology tests. For the COVID-19 susceptibility outcome, cases were also identified by review of health records (using International Classification of Disease (ICD) codes or physician notes). Fig. 1 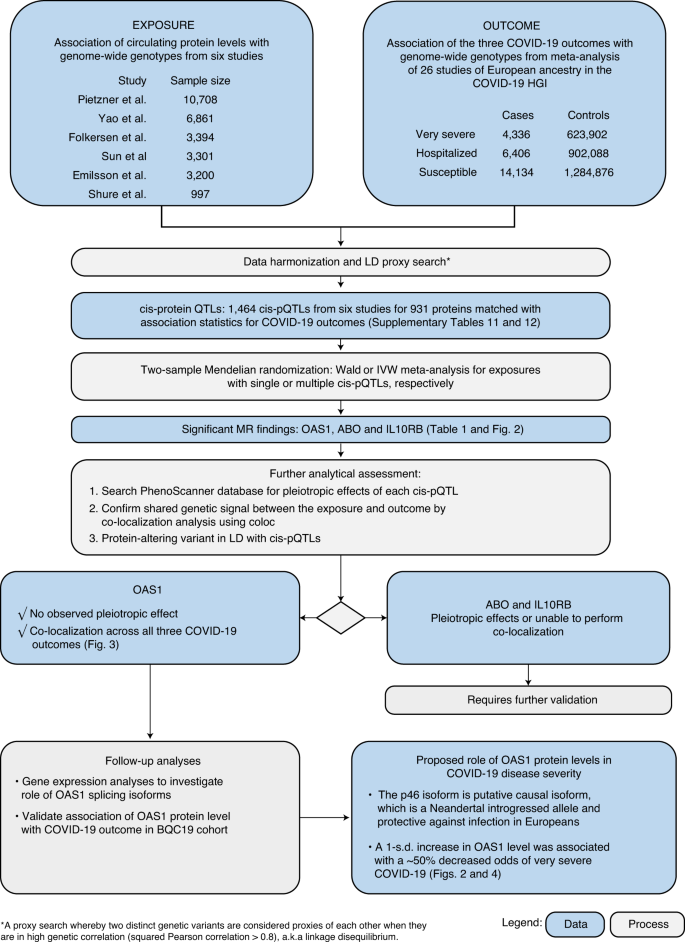 MR analyses revealed that the levels of three circulating proteins—2′–5′ oligoadenylate synthetase 1 (OAS1), interleukin-10 receptor beta subunit (IL10RB) and ABO—were associated with at least two COVID-19 outcomes after Benjamini–Hochberg false discovery rate correction (Table 1 and Supplementary Tables 1–6). Notably, increased OAS1 levels were strongly associated with protection from all three COVID-19 outcomes. Furthermore, these effect sizes were more pronounced with more severe outcomes, such that each s.d. increase in OAS1 levels was associated with decreased odds of very severe COVID-19 (OR = 0.54, 95% confidence interval (CI) 0.44–0.68, P = 7.0 × 10−8), hospitalization (OR = 0.61, 95% CI 0.51–0.73, P = 8.3 × 10−8) and susceptibility (OR = 0.78, 95% CI 0.69–0.87, P = 7.6 × 10−6) (Fig. 2a). We also identified OAS1 cis-pQTLs in Emilsson et al.16 and Pietzner et al.17, which were not included in the initial MR due to lack of genome-wide significance for their association with OAS1 levels16 or not included in their COVID-19 discovery panel17. MR analyses of using these additional cis-pQTLs yielded concordant results (Supplementary Table 7). Fig. 2: Association of circulating protein levels of OAS1, ABO and IL10RB and messenger RNA levels of OAS1 with COVID-19 outcomes from MR. 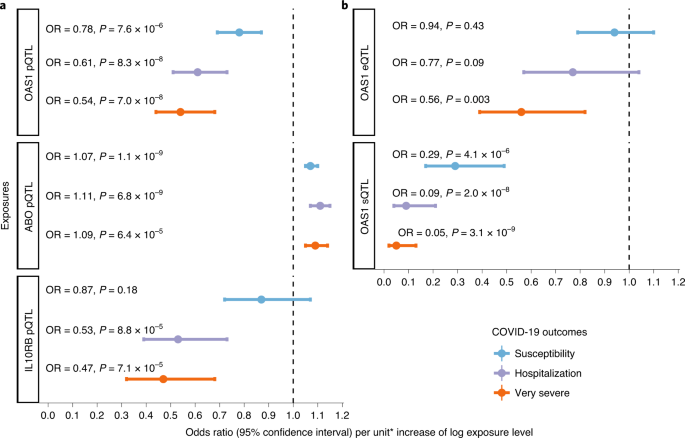 We next assessed whether the cis-pQTL for OAS1 levels (rs4767027) was associated with over 5,000 other diseases, traits or protein levels, as catalogued in PhenoScanner23. rs4767027 was not associated with any other traits or protein levels (P < 5.0 × 10−5). These findings reduce the possibility that the MR estimate of the effect of OAS1 on COVID-19 outcomes is due to horizontal pleiotropy. Finally, except for COVID-19 susceptibility, the effect of rs4767027 did not demonstrate evidence of heterogeneity across COVID-19 Host Genetics Initiative GWAS meta-analyses (Table 1). Using a cis-pQTL for IL10RB (rs2834167), we found that a 1-s.d. increase in circulating IL10RB level was associated with decreased odds of very severe COVID-19 (OR = 0.47, 95% CI 0.32–0.68, P = 7.1 × 10−5) and hospitalization (OR = 0.53, 95% CI 0.39–0.73, P = 8.8 × 10−5) but not susceptibility (Fig. 2a). Using PhenoScanner, we did not find evidence of pleiotropic effects of the cis-pQTL for IL10RB. A 1-s.d. increase in circulating ABO level was associated with increased odds of adverse COVID-19 outcomes (Table 1); however, we found that the cis-pQTL for ABO (rs505922) was strongly associated with the levels of several other proteins, suggesting potential horizontal pleiotropic effects (Supplementary Table 8). Given ABO’s known involvement in multiple physiological processes, these results were expected but highlight that MR analyses might suffer from significant bias from horizontal pleiotropy. |
|
|
|
Post by Admin on Mar 7, 2021 20:18:07 GMT
Co-localization studies To test whether confounding due to LD might have influenced the estimated effect of circulating OAS1 on COVID-19 outcomes, we tested the probability that the genetic determinants of OAS1 circulating protein level were shared with the three COVID-19 outcomes using co-localization analyses, as implemented in coloc12. The posterior probability that OAS1 levels and COVID-19 outcomes shared a single causal signal in the 1-Mb locus around the cis-pQTL, rs4767027, was 0.72 for very severe COVID-19, 0.82 for hospitalization due to COVID-19 and 0.89 for COVID-19 susceptibility (Fig. 3). This co-localization result was also replicated using OAS1 cis-pQTL identified by Pietzner et al.17 (Supplementary Table 7). This suggests that there is likely a single shared causal signal for OAS1 circulating protein levels and COVID-19 outcomes. Fig. 3: Co-localization of the genetic determinants of OAS1 plasma protein levels and COVID-19 outcomes. 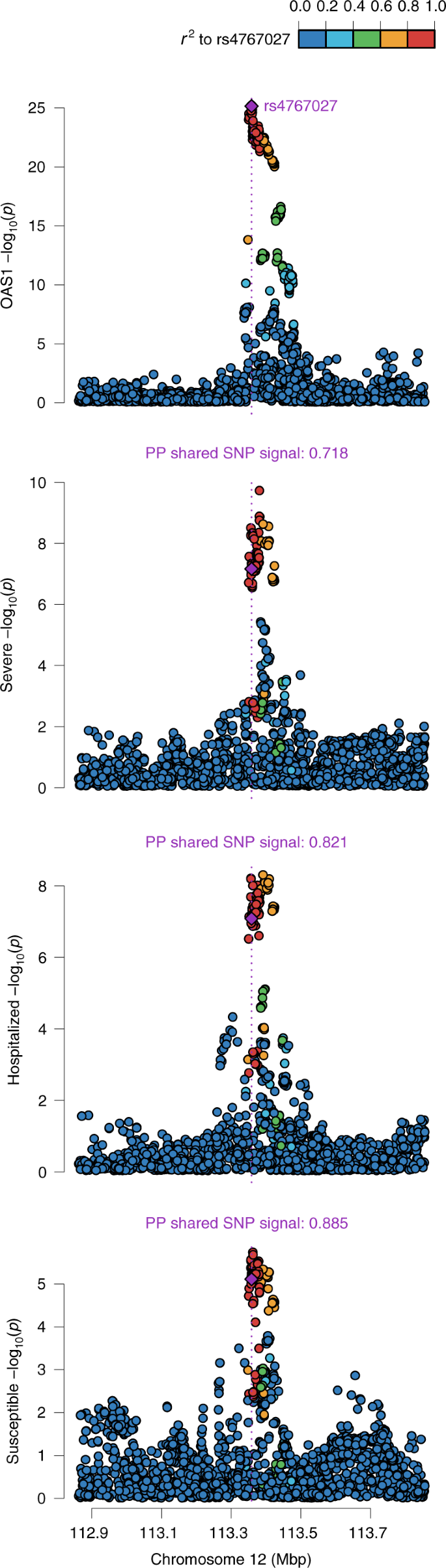 Co-localization of genetic signal for OAS1 levels (top plot) and COVID-19 outcomes (three bottom plots) in the 1-Mb region around OAS1 pQTL rs4767027; color shows SNPs in the region in LD (r2) with rs4767027 (purple). The posterior probability (PP) of a shared single signal between OAS1 levels and the three COVID-19 outcomes was estimated by coloc. Co-localization of ABO levels and different COVID-19 outcomes also showed co-localization between ABO level and different COVID-19 outcomes (posterior probability of single shared signal = 0.90, 0.98 and 1 for ABO level and very severe COVID-19, hospitalization due to COVID-19 and susceptibility, respectively) (Extended Data Fig. 1). We were unable to perform co-localization analyses for IL10RB due to a lack of genome-wide summary-level data from the original proteomic GWAS16. Aptamer-binding effects Protein-altering variants (PAVs)15 might influence binding of affinity agents, such as aptamers or antibodies, that are used to quantify protein levels. We, thus, assessed if the cis-pQTLs for the MR-prioritized proteins were PAVs or in LD (r2 > 0.8) with PAVs. rs2834167 (IL10RB) is a nonsense variant and could, therefore, be subject to potential binding effects. rs505922 (ABO) is not in LD with known missense variants. rs4767027 (OAS1) is an intronic variant, which is in LD with a missense variant rs2660 (r2 = 1) in European ancestry. However, because expression studies derived from RNA sequencing are not subject to potential effects of missense variants that could influence aptamer binding, we next explored whether rs4767027 also influences OAS1 expression and/or splicing. sQTL and eQTL studies for OAS genes sQTLs are genetic variants that influence the transcription of different isoforms of a protein. The aptamer that targets OAS1 was developed against a synthetic protein comprising the amino acid sequence 1–364 of NP002525.2, which is common to the two major OAS1 isoforms: p46 and p42. Hence, the aptamer might identify both or either isoforms. rs10774671 is a known sQTL for OAS1 that induces alternate splicing and creates p46 and p42 isoforms. Most present-day individuals of European ancestry carry the alternative variant (rs10774671-A). The ancestral variant (rs10774671-G) is the major allele in African populations and became fixed in Neanderthal and Denisovan genomes24,25. However, the ancestral variant, with its increased expression of the p46 isoform, was reintroduced into the European population via gene flow from Neanderthals26. Previous analyses suggest that individuals with either the GG or GA genotype at rs10774671 express higher amounts of p46 (ref. 26), which is also the predominant isoform found in circulating blood27. Differences in antiviral activity have been observed between isoforms, with p46 being more active in certain viral infections28. Interestingly, the OAS1 pQTL rs4767027 is in high LD (r2 = 0.97) with rs10774671 (ref. 26) in European populations. Functional studies support that the G allele at rs10774671 increases expression of the p46 isoform but decreases expression of the p42 isoform27. This G allele at the sQTL rs10774671 reflects the T allele at pQTL rs4767027, which itself is associated with higher measured OAS1 levels and reduced odds of COVID-19 severity and susceptibility. These separate lines of evidence suggest that OAS1 levels, as measured by the SomaScan platform, predominantly identify the p46 isoform, which might protect against COVID-19 outcomes. Undertaking MR studies of OAS1 splicing, we found that increased expression of the p46 isoform (as defined by normalized read counts of the intron cluster defined by LeafCutter29,30) was associated with reduced odds of COVID-19 outcomes (OR = 0.29, 95% CI 0.17–0.49, P = 4.1 × 10−6 for susceptibility, OR = 0.09, 95% CI 0.04–0.21, P = 2.0 × 10−8 for hospitalization and OR = 0.05, 95% CI 0.02–0.13, P = 3.1 × 10−9 for very severe COVID-19) (Fig. 2b). Co-localization analyses also supported a shared causal signal among the sQTL for OAS1, the pQTL and COVID-19 outcomes (Extended Data Fig. 2). Interestingly, the co-localization analyses supported a stronger probability of a shared signal with the sQTL than the pQTL, suggesting that the p46 isoform might be the driver of the association of OAS1 levels with COVID-19 outcomes. Next, we tested, using eQTL MR analyses, whether increased expression of OAS1 levels, without respect to isoform, was associated with COVID-19 outcomes. We identified an eQTL for total OAS1, rs10744785, from GTEx v8 (ref. 31). Total OAS1 expression levels were not associated with COVID-19 susceptibility and hospitalization (Fig. 2b). We also found that increased OAS3 expression in whole blood was positively associated with COVID-19 outcomes in MR analyses and support for co-localization of their genetic signal (Extended Data Fig. 3 nd Supplementary Table 9). |
|
|
|
Post by Admin on Mar 7, 2021 22:32:45 GMT
Association of measured OAS1 protein level with COVID-19 outcomes Because MR studies were derived from protein levels measured in a non-infected state, we tested the hypothesis that increased OAS1 protein levels in a non-infected state would be associated with reduced odds of COVID-19 outcomes. To do so, we undertook a case–control study, measuring OAS1 protein levels using the SomaScan platform in 1,039 longitudinal samples from 399 patients who tested positive for SARS-CoV-2 by polymerase chain reaction (PCR) that were collected at multiple time points during their COVID-19 infection and 105 individuals who presented with COVID-19 symptoms but had negative SARS-CoV-2 PCR nasal swabs from the Biobanque Quebecoise de la COVID-19 cohort (www.BQC19.ca). Individuals who had undergone nasal swabs for SARS-CoV-2 infection were recruited prospectively (Table 2). We defined non-infectious samples as those collected from convalescent patients with SARS-CoV-2 at least 31 d after onset of their symptoms (n = 115) or samples collected from patients negative for SARS-CoV-2 by PCR (n = 105). We also measured OAS1 levels in individuals with samples from patients positive for SARS-CoV-2 <14 d after symptom onset (n = 313), which showed increased OAS1 levels during infection (Extended Data Figs. 4–6). OAS1 levels are not associated with age and sex in non-infectious samples (Extended Data Fig. 7). After sample quality control (Methods), 308 patients with at least one sample collected during infection, 113 patients with at least one sample collected during a non-infectious state and 103 COVID-19-negative controls were included in the analyses (Extended Data Fig. 8). To test whether OAS1 levels in a non-infectious state were associated with COVID-19 outcomes, we undertook logistic regression controlling for age, sex, age*age, plate, recruitment center and sample processing time. OAS1 levels were log-transformed and standardized to match the transformation procedure of the MR study. We found that, in the non-infectious samples, each s.d. increase in OAS1 levels on the log-transformed scale was associated with reduced odds of COVID-19 outcomes (OR = 0.20, 95% CI 0.08–0.53, P = 0.001 for very severe COVID-19; OR = 0.46, 95% CI 0.28–0.76, P = 0.002 for hospitalization; and OR = 0.69, 95% CI 0.49–0.98, P = 0.04 for susceptibility) (Fig. 4, Extended Data Fig. 9 and Supplementary Table 10). These results are consistent with our findings from MR, where increased circulating OAS1 levels in a non-infectious state were associated with protection against all of these adverse COVID-19 outcomes. Fig. 4: Association of OAS1 levels with COVID-19 outcomes from the case–control study in BQC19. 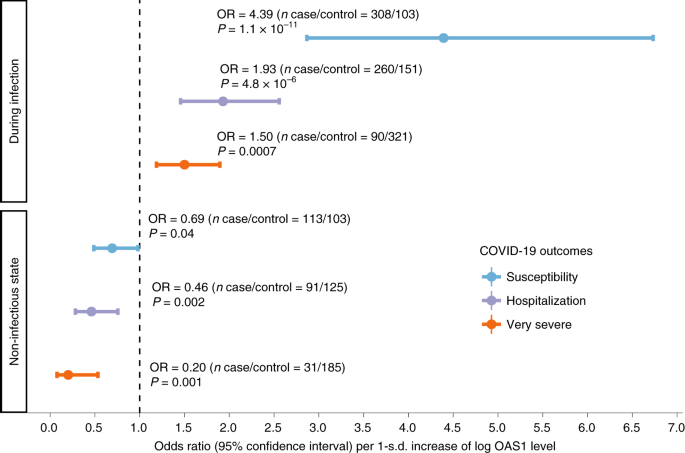 Forest plot showing ORs and 95% CIs from logistic regression analyses (two sided). P values are unadjusted. During infection: patient samples that were collected within 14 d from the date of symptom onset. For individuals with two or more samples collected within 14 d of symptom onset, the earliest time point was used. Non-infectious state: patient samples that were collected at least 31 d from the date of symptom onset. For individuals with two or more samples collected at different time points at least 31 d from symptom onset, the latest time point was used. Additional information is also described in Supplementary Table 10. In samples drawn during active infection, we found that increased OAS1 levels were associated with increased odds of adverse COVID-19 outcomes (OR = 1.50, 95% CI 1.19–1.90, P = 0.0007 for very severe COVID-19; OR = 1.93, 95% CI 1.46–2.56, P = 4.8 × 10−6 for hospitalization; and OR = 4.39, 95% CI 2.87–6.73, P = 1.09 × 10−11 for susceptibility) (Fig. 4). Taken together, these findings suggest that increased OAS1 levels in a non-infectious state are associated with better COVID-19 outcomes, and that, during infection, SARS-CoV-2 exposure likely causes OAS1 levels to increase, as interferon pathways are stimulated, which are known to increase OAS1 levels32. |
|

