|
|
Post by Admin on Oct 17, 2023 5:57:37 GMT
Local ancestry estimation We carried out a supervised population structure analysis by applying LAI with RFMix using a reference panel of haplotypes from Africa, Europe and America (Methods). Supplementary Fig. 11 shows local ancestry at segments genome-wide for 12 representative MCPS individuals estimated from the LAI results. Figure 3 shows population distributions of LAI-based ancestry proportion estimates, including Indigenous American ancestry from five geographical regions within Mexico. Overall, we estimated that 66.0% of autosomal ancestry was attributable to Indigenous Mexican populations, with the majority coming from central Mexico (35.6%). Southern Mexico and southeastern Mexico accounted for 15.9% and 11.8%, respectively, with much smaller amounts of ancestry attributable to northern Mexico (1.6%) and northwestern Mexico (1.1%). In addition, 2.9% and 31.1% of ancestry were attributable to African and European populations, respectively. We observed that MCPS individuals with the most Indigenous Mexican ancestry seemed to have a greater relative contribution from Indigenous populations from southern Mexico (that is, from the states of Oaxaca and Veracruz) (Supplementary Fig. 12). Moreover, lower amounts of Indigenous Mexican ancestry and higher amounts of European ancestry were observed in Coyoacán than in Iztapalapa, a result consistent with the sociodemographic characteristics of these districts. Fig. 3: Global ancestry proportions estimated from LAI. 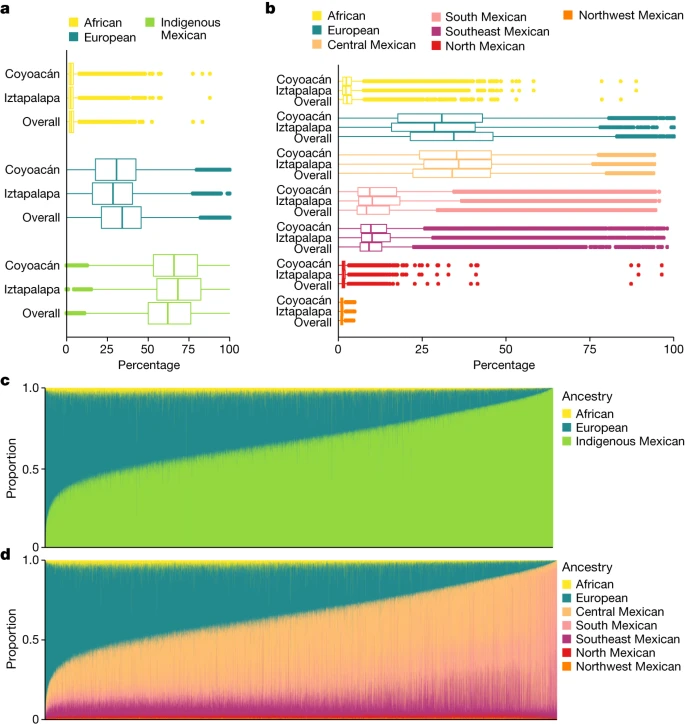 a,b, Distributions of LAI-based global ancestry proportions for n = 138,511 MCPS individuals from a 7-way analysis (b) and reduced to 3 continental populations (a). c,d, Stacked bar plots of three-way (c) and seven-way (d) global ancestry proportions for n = 138,511 MCPS individuals. Using 3,595 parent couples inferred from the genetic relatedness analysis, we observed significant correlation in ancestry between partner pairs (Supplementary Fig. 13), as has been observed in other studies in admixed populations18. Education and district explained between 0.5 and 5% of the variation in ancestry, whereas spousal ancestry explained between 15 and 26% of the variation in ancestry. This result suggests that genomic ancestry is a better predictor of the ancestry of partners than these sociodemographic factors. Extended Data Fig. 4 shows the proportion of ancestry across each chromosome from a three-way LAI analysis, and Supplementary Fig. 14 shows per-ancestry tests for departures from genome-wide ancestry proportions (Methods). This result highlighted an excess of African ancestry in and around the MHC locus on chromosome 6 (African 17.3%, P = 2.9 × 10–14; Supplementary Fig. 15), a result consistent with previous observations19. An additional signal on chromosome 15 showed increased European ancestry of 35.2% at position 48.38 Mb (P = 3.8 × 10–8) and spanned a region between 45.09 Mb and 52.31 Mb in and around SLC24A5, a gene with known function in human skin pigmentation. Variant rs1426654 in SLC24A5 explains roughly one-third of the variation in pigmentation between Europeans and West Africans, probably being under selection in Europeans20. We also observed ancestry proportions on chromosome X that exhibited increased levels of Indigenous Mexican ancestry compared with the autosomes (African 3.2%, Indigenous Mexican 73.8%, European 22.7%), a finding consistent with an imbalance of male and female contributions to admixture. Using a simplified population mixture event model21,22 that best fit the observed chromosome X ancestry proportions, we estimated that the proportion of Indigenous Mexican ancestry explained by female contribution was 71.3%. By contrast, for Europeans, the female contribution accounted for 7.5% (Supplementary Table 28). |
|
|
|
Post by Admin on Oct 18, 2023 6:01:29 GMT
Homozygosity
Increased levels of homozygosity were indicated by both the relatedness analysis, which highlighted parent–offspring pairs with increased levels of sharing two alleles IBD genome-wide (Fig. 1a), and the exome variant survey, which highlighted high counts of homozygous pLOF variants compared with the UK Biobank exome dataset (Supplementary Table 4). We assessed homozygosity by estimating ROH from the phased array dataset using hap-IBD (Methods), which produced a mean homozygosity of 0.34% for all MCPS individuals. There were 60,722 MCPS participants (43.9%) who had at least one ROH segment 4 centiMorgan (cM) or longer, for which the mean homozygosity was 0.78% (Supplementary Table 29 and Extended Data Fig. 5). By comparison, for the UK Biobank data, the mean homozygosity was 0.07%, and 0.59% among the 55,206 (11.3%) participants who had at least one ROH segment ≥4 cM.
We observed that the total length and number of ROH segments were positively correlated with the proportion of ancestry native to Mexico (Supplementary Fig. 16). Overall, 79.0% of ROH segments could be assigned to Indigenous Mexican ancestry when overlaid with inferred local ancestry (Methods), which exceeded the 66.3% average amount of Indigenous Mexican ancestry in the sample. Conversely, we observed a depleted proportion of European and African ancestry in ROH segments (19.10% and 1.9%, respectively) compared with the average amount in the sample (30.2% and 3.5%, respectively), which was consistent with previous findings23.
The mean number of rare homozygous pLOFs (rhLOF; allele frequency of <0.1%) and the proportion of rhLOFs in ROH correlated with the proportion of the genome in ROH segments (Supplementary Fig. 17). We identified 3,763 rhLOF genotypes at 2,646 variants in 2,169 different protein-coding genes in 3,519 individuals, and 52.2% of these were found within ROH segments. Consistent with the rate of rhLOF variants and assignment of ROH segments to Indigenous American ancestry (Supplementary Table 4), segments of Indigenous Mexican ancestry accounted for 62.6% of rhLOFs, a result indicative of an ancestry-specific trend (Supplementary Table 30).
|
|
|
|
Post by Admin on Oct 19, 2023 6:59:45 GMT
An MCPS imputation reference panel We created a phased haplotype imputation reference panel (MCPS10k) from the 9,950 WGS individuals utilizing sequencing reads, pedigrees and a phased array haplotype scaffold (Methods). Using the WGS trios, we estimated that haplotypes were phased with a switch error rate of 0.0024 (Methods and Supplementary Fig. 18) and observed that the switch error rate depended on ancestry proportion (Supplementary Fig. 19). We assessed the utility of the MCPS10k reference panel for genotype imputation by imputing chromosome 2 using the phased array dataset of 67,079 MCPS individuals not included in the reference panel and pruned for relationships up to the first degree. For comparison, we also imputed the MCPS dataset using the diverse TOPMed reference panel that includes 47,159 European, 24,267 African and 17,085 admixed American genomes (Methods). MCPS10k and TOPMed imputation produced a set of 9,801,290 and 9,437,266 variants, respectively, on chromosome 2, with an imputation information score of >0.3. However, the information scores (a well-calibrated measure of accuracy) for an overlapping set of 6,473,872 variants were generally higher using MCPS10k than TOPMed for MAF bins greater than 0.01% (Extended Data Fig. 6). We compared the MCPS10k and TOPMed imputed genotypes to the exome-sequencing data at 128,728 sites on chromosome 2. Figure 4 shows the results of the imputation accuracy stratified by allele frequency, reference panel and degree of Indigenous Mexican ancestry (defined as two groupings with individuals split above and below the median proportion of Indigenous Mexican ancestry). The results showed that MCPS10k had a comparable performance with TOPMed across the entire frequency range. However, the MCPS10k panel provided the greatest imputation benefits for the samples with high proportions of Indigenous Mexican ancestry. Fig. 4: Imputation accuracy using the MCPS10k and TOPMed imputation panels. 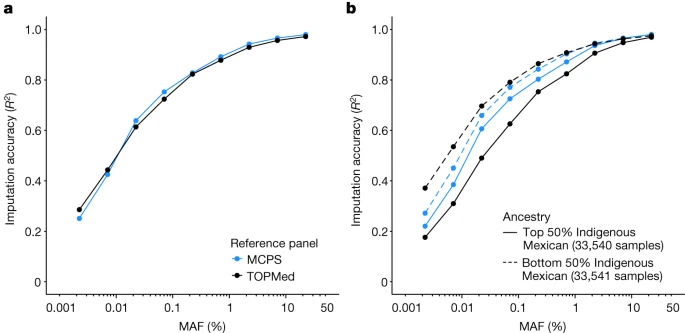 a,b, Accuracy was measured using the R2 between the imputed variants and 128,728 variants measured using exome sequencing on chromosome 2 in 67,079 MCPS samples not in (or related to) the MCPS reference panel samples. Results are stratified by allele frequency (x axis on log10 scale), reference panel and into two populations (top and bottom 50% of Indigenous Mexican ancestry shown by solid and dashed lines). a, Results for all samples. b, Results stratified by the amount of Indigenous Mexican estimated in each sample. Full size imageFinally, we assessed the imputation performance in Mexican Americans from Los Angeles (MXL) in 1000 Genomes and found that TOPMed provided improved imputation performance compared with MCPS10k (Supplementary Figs. 20 and 21). This result is probably driven by MXL samples having substantially higher European ancestry and less ancestry from central, southern and southeast Mexico than in the MCPS cohort (Supplementary Fig. 22). Similarly, the TOPMed panel provided the best performance in individuals with Peruvian ancestry from Lima (PEL), Colombian ancestry from Medellin (CLM) and Puerto Rican ancestry from Puerto Rico (PUR) from the 1000 Genomes study compared with MCPS10k (Supplementary Figs. 20 and 21). These results emphasize the value of closely matching the ancestry of imputation reference panels to the samples being studied. Although our panel provided improved imputation for individuals of Mesoamerican Mexican ancestry, additional panels may be required to provide similar benefits for other Latin American populations with admixture from different Indigenous American ancestral populations. |
|
|
|
Post by Admin on Nov 16, 2023 21:45:26 GMT
Genetic diversity of the melanocortin-1 receptor in an admixed population of Rio de Janeiro: Structural and functional impacts of Cys35Tyr variant Abstract The melanocortin-1 receptor (MC1R) is one of the key proteins involved in the regulation of melanin production and several polymorphisms have been associated with different phenotypes of skin and hair color in human and nonhuman species. Most of the knowledge is centered on more homogeneous populations and studies involving an admixed group of people should be encouraged due to the great importance of understanding the human color variation. This work evaluates the MC1R diversity and the possible impacts of MC1R variants in an admixed sample population of Rio de Janeiro, Brazil, which is a product of Native American, African, and European miscegenation. Sequencing of complete coding region and part of the 3´UTR of MC1R gene identified 31 variants including one insertion and three novel synonymous substitutions in sample population grouped according to skin, hair and eye pigmentation levels. In nonmetric multidimensional scaling analysis (NMDS), three main clusters were identified, in which the Brazilian dark skin group remained in the African cluster whereas the intermediate and the light skin color phenotype in the European one. None gathered with Asians since their immigration to Brazil was a recent event. In silico analyses demonstrated that Cys35Tyr, Ile155Thr and Pro256Ser, found in our population, have a negative effect on receptor function probably due to changes on the receptor structure. Notably, Cys35Tyr mutation could potentially impair agonist binding. Altogether, this work contributes to the understanding of the genetic background of color variation on an admixed population and gives insights into the damaging effects of MC1R variants. Introduction Color variation is a complex trait orchestrated by elaborate signaling networks that are shaped by a set of genetic variants and epigenetic modifications. One of the most well studied genes governing the process of melanogenesis is the melanocortin-1 receptor (MC1R) that belongs to the family of G protein-coupled receptors (GPCRs) and encodes a protein with 317 amino acids composed of seven transmembrane domains (TM) with a high affinity for melanocyte stimulating hormone (MSH). MC1R is a highly polymorphic gene and it has been related to pigmentary as well as to non-pigmentary functions, including DNA repair (10.3390/genes12071093). MC1R plays a key role in skin protection against damaging ultraviolet radiation by regulating eumelanin production [1]. Preferentially present in melanocytes, the receptor activated by its agonists favors the synthesis of brown/black eumelanin instead of (yellow/red) pheomelanin, which predominates under condition of none or low signal transduction [2, 3]. The balance of eumelanin/pheomelanin production is a sophisticated mechanism to avoid the harmful consequences of ultraviolet (UV) exposure, not only in terms of DNA mutagenesis but mainly in maintaining folate integrity and vitamin D production [4]. For that reason, MC1R undergoes a directional selective pressure according to the geographic region reflected on the skin color: the more UV incidence, the darker the skin [4]. MC1R-defective individuals, because of their tendency to be undermelanized, they should accumulate more UV mutagenesis over time and would therefore be at higher risk for melanoma as a result [5, 6]. MC1R is overexpressed on the cell surface of most human melanomas making MC1R a valuable marker of these tumors. Although findings support association of genetic variants of MC1R of hair color and Parkinson’s disease (PD) risk [7] it is still controversial that variants of MC1R account for co-occurrence of PD and malignant melanoma [8]. MC1R is under strong evolutionary constraint in African origin since any underproduction of eumelanin appears to be deleterious [9]. As ancient humans migrated out of Africa towards low-UV regions in Europe or Asia, beneficial mutations have swept to fixation promoting convergent skin lightening [4, 10]. Most notably in European populations, many polymorphisms have been associated with fair skin phenotype, red hair color (RHC), presence of freckles as well as increasing predisposition of skin cancer [6, 11, 12]. The MC1R polymorphisms are best known for discriminating red hair from non-red hair phenotype due to mutations that impair the function of the receptor, disrupting the production of dark melanin [11]. The so-called strong alleles “R” for RHC have high penetrance for red hair and fair skin: Asp84Glu (rs1805006), Arg142His (rs11547464), Arg151Cys (rs1805007), Arg160Trp (rs1805008) and Asp294His (rs1805009). Some variants are predicted as damaging mutations such as Ser83Pro (rs34474212) and Pro256Ser (rs200215218) and also, variant for the loss-of-function Cys35Tyr (rs779504604) [13, 14]. www.ncbi.nlm.nih.gov/pmc/articles/PMC9032367/ |
|
|
|
Post by Admin on Nov 17, 2023 21:08:37 GMT
Initially, studies on MC1R were based on individuals of unmixed origin who tend to have low intrapopulation skin color variation. However, few studies have been conducted in order to understand the genetic diversity and the phenotypic associations that comprise the intermediate levels between low and high melanin production, requiring further investigation in admixed populations [15–17]. Brazil is considered one of the most genetically diversified and highly admixed populations [18]. The process of colonization, undertaken firstly by Europeans with the contribution of Amerindians as native population, black Africans slaves and more recently Asians, has resulted in a broad range of color variations, more precisely in skin pigmentation [19]. In addition, the ethnic proportions and contributions of these parental populations (Amerindians, Africans and Europeans), observed in the Rio de Janeiro (RJ) population sample showed higher prevalence of European ancestry (around 60%) followed by African (25%) and native American (11%) [20], nevertheless, the distribution of skin color exhibits a wide range variation of phenotypes from low to high melanin content [21–23].
The present work approaches the genetic diversity of melanocortin-1 receptor in an admixed population with distinct color of skin, hair and eye. We sequenced the entire coding region and part of the 3´UTR of MC1R in a diverse population in terms of pigmentation levels and evaluated the phenotypic distribution related to skin color among the RJ population and those from the 1000 Genomes consortium [24], representing the parental contribution from colonization of Brazil to the present. Analysis based on population distribution, light and intermediate skin remains gathered to Europeans, despite the intermediate matrilineal ancestry of majority African origin. We also addressed the impact of distinct SNPs on the function and structure of the receptor using in silico methods. Variants associated with five point mutations (Cys35Tyr, Ile155Thr, Pro256Ser, Val156Leu and Phe196Leu), predicted as damaging mutations, were analyzed in silico to understand their structural effects and predict their functional roles on MC1R. Cys35Tyr is particularly significant due to the disruption of a critical disulfide bond and overall protein destabilization. Molecular dynamics simulations evidenced the mutation effect by structural conformational changes that potentially impair agonist binding. This work contributed to the understanding of the intermediate spectrum of color variation, and the functional implications of MC1R variants.
|
|

