Post by Admin on Jul 19, 2021 20:20:59 GMT
Insights Into the Formation, Genetic Structure, and Phylogenetic Relationship of Northern Han Chinese
Pengyu Chen1,2†, Jian Wu1,2†, Li Luo1,2†, Hongyan Gao1,2, Mengge Wang3, Xing Zou3, Yingxiang Li4, Gang Chen4, Haibo Luo2, Limei Yu5, Yanyan Han6, Fuquan Jia7* and Guanglin He3*
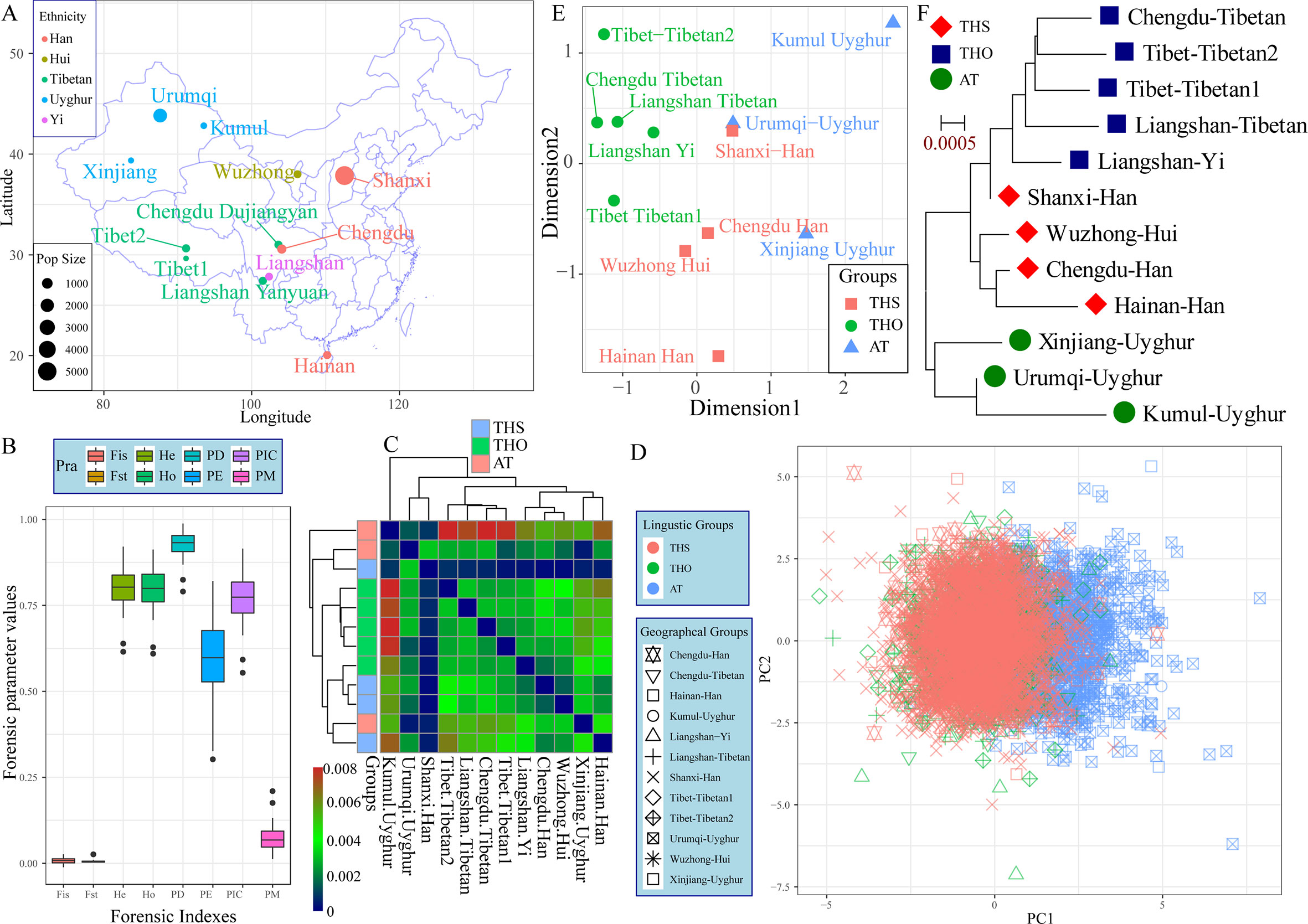
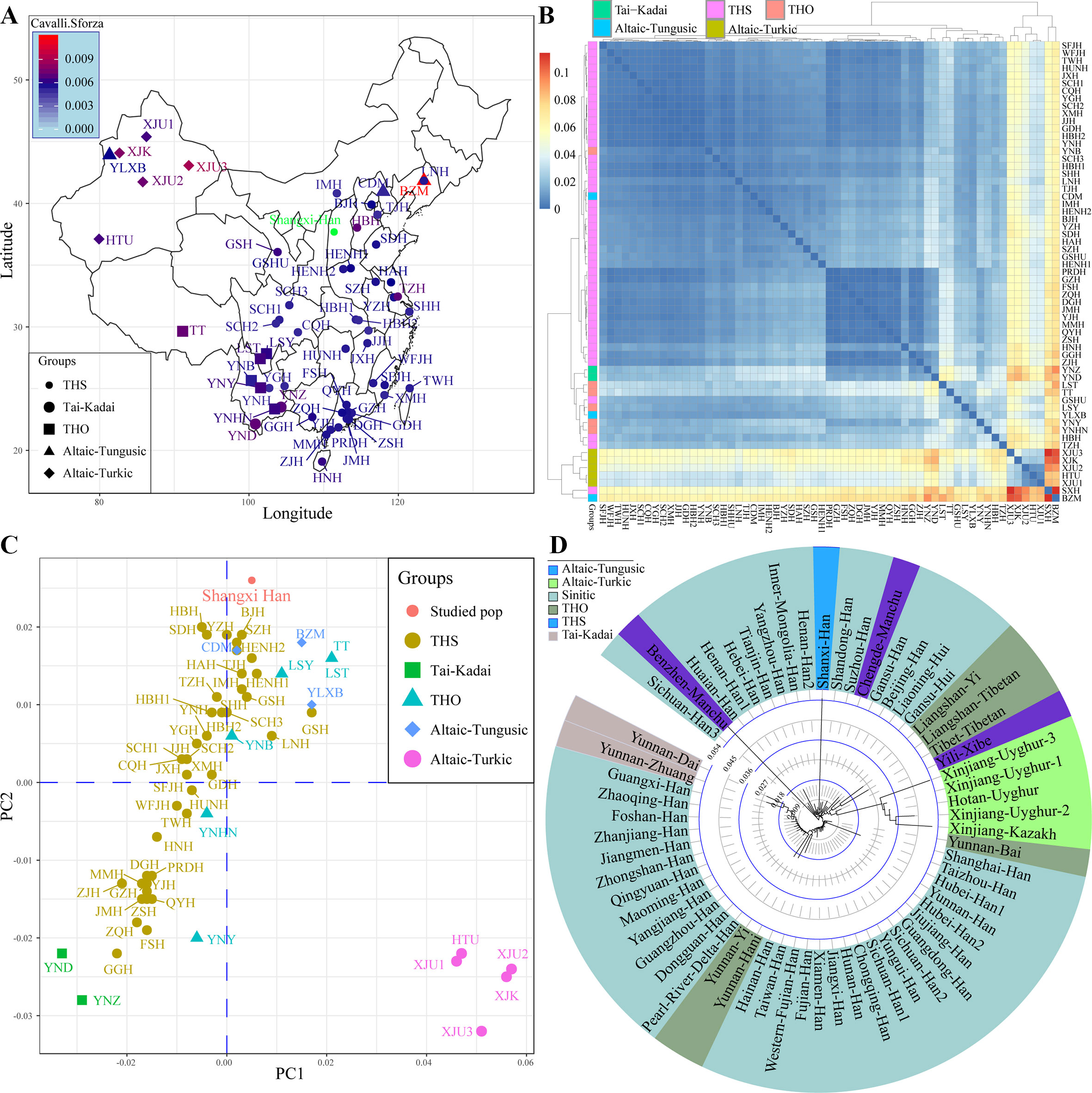
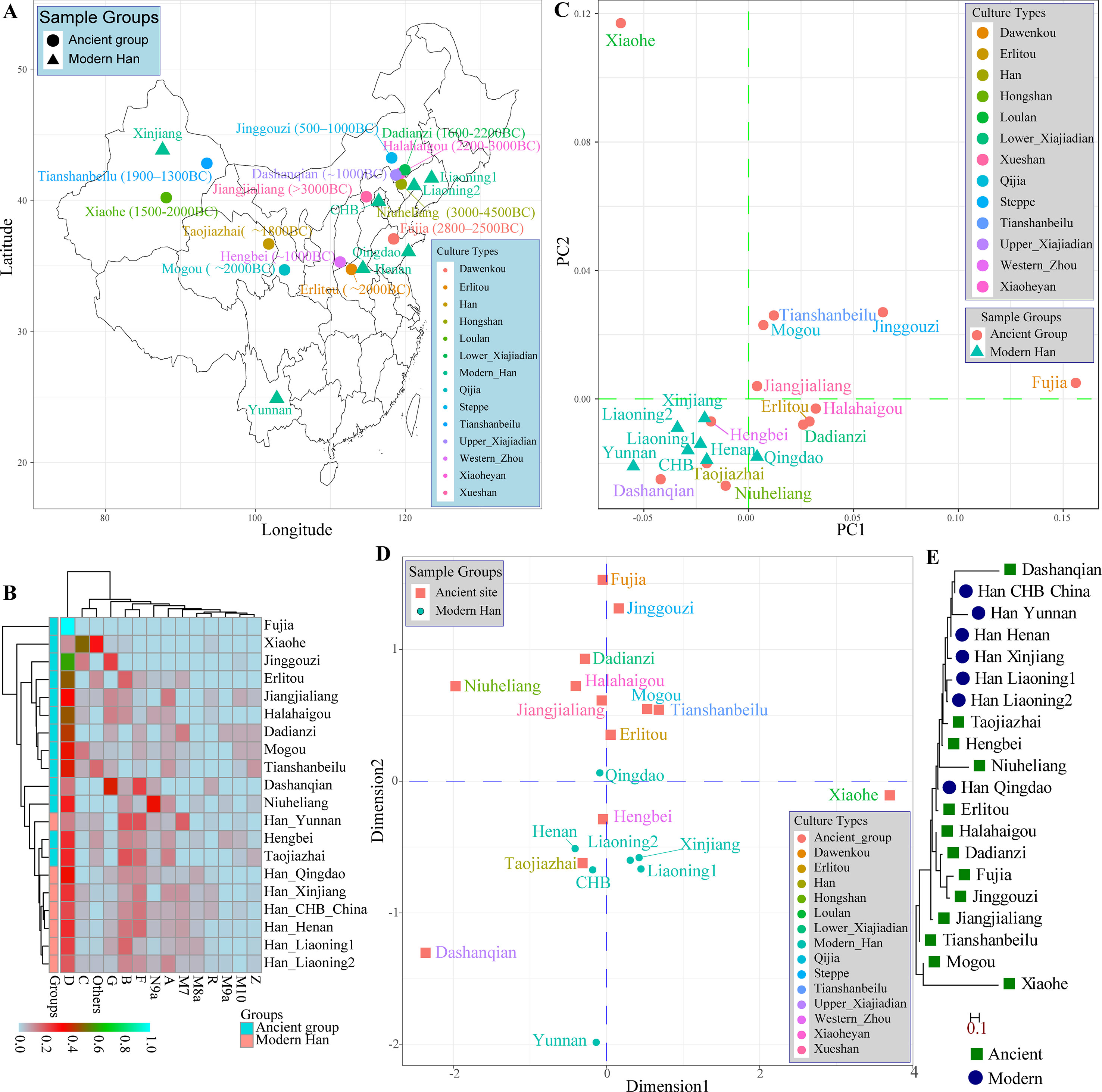
Modern East Asians derived from the admixture of aborigines and incoming farmers expanding from Yellow and Yangtze River Basins. Distinct genetic differentiation and subsequent admixture between Northeast Asians and Southeast Asians subsequently evidenced by the mitochondrial DNA, Y-chromosomal variations, and autosomal SNPs. Recently, population geneticists have paid more attention to the genetic polymorphisms and background of southern-Han Chinese and southern native populations. The genetic legacy of northern-Han remains uncharacterized. Thus, we performed this comprehensive population genetic analyses of modern and ancient genetic variations aiming to yield new insight into the formation of modern Han, and the genetic ancestry and phylogenetic relationship of the northern-Han Chinese population. We first genotyped 25 forensic associated markers in 3,089 northern-Han Chinese individuals using the new-generation of the Huaxia Platinum System. And then we performed the first meta-analysis focused on the genetic affinity between Asian Neolithic∼Iron Age ancients and modern northern-Han Chinese by combining mitochondrial variations in 417 ancient individuals from 13 different archeological sites and 812 modern individuals, as well as Y-chromosomal variations in 114 ancient individuals from 12 Neolithic∼Iron Age sites and 2,810 modern subjects. We finally genotyped 643,897 genome-wide nucleotide polymorphisms (SNPs) in 20 Shanxi Han individuals and combined with 1,927 modern humans and 40 Eurasian ancient genomes to explore the genetic structure and admixture of northern-Han Chinese. We addressed genetic legacy, population structure and phylogenetic relationship of northern-Han Chinese via various analyses. Our population genetic results from five different reference datasets indicated that Shanxi Han shares a closer phylogenetic relationship with northern-neighbors and southern ethnically close groups than with Uyghur and Tibetan. Genome-wide variations revealed that modern northern-Han derived their ancestry from Yakut-related population (25.2%) and She-related population (74.8%). Summarily, the genetic mixing that led to the emergence of a Han Chinese ethnicity occurred at a very early period, probably in Neolithic times, and this mixing involved an ancient Tibeto-Burman population and a local pre-Sinitic population, which may have been linguistically Altaic.
Introduction
Han Chinese, with a total population size circa 1.4 billion, is the world’s largest ethnic group and dominant ethnicity in China and Singapore. The origin of the Han Chinese population, genetic relationship with adjacent groups and past migratory pattern and admixture history have gained considerable attention from scientists working in the anthropology, linguistics, history, population and forensic genetics (Zhao et al., 2011; Gao et al., 2015; Zhao et al., 2015b; Li et al., 2017; Nothnagel et al., 2017; Zhang et al., 2017b; Chiang et al., 2018). Archaeological and anthropological evidence showed that human occupation in East Asia has experienced archaic hominin extinction, genetic introgression between early anatomically modern human and Denisovan or Neanderthals, the transformation from hunting–gathering to agriculture, massive admixture and migratory history with ethnolinguistically diverse populations in the past 50–100 thousand years (Nielsen et al., 2017). Expansions of the maternally-inherited mitochondrial DNA (mtDNA) and paternally-inherited Y-chromosome haplogroup lineages indicated that ethnically different East Asians derived from southeastern groups and experienced south-to-north migrations driven by a variety of evolutionary mechanisms (Su et al., 1999; Yao et al., 2002). Besides, social practices, including subsistence strategies, residence patterns, and agricultural expansion, play an indispensable role in shaping the patterns of Chinese populations (Nielsen et al., 2017). Ancient mitochondrial and Y-chromosomal DNA studies in East Asian Neolithic∼Iron Age populations have drastically increased in past decades (Cui et al., 2010; Li et al., 2010; Li et al., 2011; Zhao et al., 2011; Wang et al., 2012; Cui et al., 2013; Zhao et al., 2014; Dong et al., 2015; Gao et al., 2015; Li et al., 2015; Zhao et al., 2015b; Li et al., 2017; Zhang et al., 2017b; Li et al., 2018), however, how the peopling and settlement history of Neolithic populations influence the origin, expansion, and migration of the Han Chinese population is still unclear.
Physical anthropological investigation of somatometric and nonmetric features revealed that a significant difference exists between northern-Han Chinese and southern-Han Chinese (Sanchez-Mazas et al., 2011). Subsequently, Chu et al. (1998) found genetic evidence to support the distinction between southern and northern populations. Phylogeographic or genetic differentiation between northern-Han and southern-Han have been also evidenced by Yao et al. (2002) using mitochondrial DNA, Wen et al. (2004) using combined testing Y-chromosome and mitochondrial DNA variations, and Chen et al. (2009) and Xu et al. (2009) using high-density genotype data. Our previous study has investigated the genetic polymorphisms, forensic features and genetic relationship of currently widely-used autosomal short tandem repeats (STRs) in the southern-Han Chinese residing in the Pearl River Delta (He et al., 2018d). Thus, reconstructing the forensic reference database, estimating the forensic allele frequency and parameters and dissecting the genetic relationship of this genetically diverse northern-Han Chinese population are very necessary and urgent.
STR, also called as microsatellite, is one of the extraordinary mutated genetic markers, is widely existed human autosomal, X-chromosome and Y-chromosome genomes (Ge et al., 2014). This length polymorphism marker is generated by the slippage synthesis of simple sequence (2–8bp) (Schlotterer and Tautz, 1992). STRs located on the no-recombining region of Y-chromosome are the best candidates for forensic pedigree searches and identifying the perpetrator in the sexual crime or rape cases, and X-chromosomal STRs are best suitable for applications in the deficiency and incestuous cases (He et al., 2017d; Chen et al., 2018). Autosomal STR genotyping is the gold standard in the routine forensic cases. Nowadays, all organizations or countries optimized their accepted STR panels to improve the international collaboration, such as the expanded CODIS core loci, extended European standard set (ESS-extended). Huaxia Platinum System (Thermo Fisher Scientific) was integrated all twenty expanded CODIS core loci, additional STRs included in the Chinese National Database and two gender determination loci (He et al., 2018b). Single nucleotide polymorphisms (SNPs), with the number over 84.7 million in the human genome, are the best candidate to explore the detailed processes of human origin, migration, evolution, adaptation and admixture (Genomes Project et al., 2015).
Although Y-chromosomal and X-chromosomal variations of the northern-Han Chinese population have been investigated and reported (He et al., 2017d; Chen et al., 2018). Autosomal STR allele distribution of this new-generation of the Huaxia Platinum Amplification System with regard to forensic statistical features has not previously been investigated. Besides, the population genetic structure and admixture history of northern-Han via high-density genetic markers are unclear. Thus, we genotyped and analyzed 23 autosomal STRs in 3,089 unrelated Han Chinese individuals and 643,897 SNPs in 20 Hans residing in Shanxi Province. Shanxi Province is between 34°34′-40°44′ north latitude and 110°14′-114°33′ east longitude, which stretches about a total area of 156,700 km2 from the Yellow River in the west and south to the Taihang Mountain in the east and the Great Wall in the north. This area is bounded by the Shaanxi, Inner Mongolia, Hebei, Henan, and other Provinces. Archaeological, anthropological and genetic evidence from Hengbei site consistently considered that Han Chinese is originated from Shanxi and neighboring regions, also called the Central Plain (Zhao et al., 2015b). And then Han Chinese population migrated southward with the Han-associated culture (Demic diffusion) and admixed with southern Chinese natives and formed the current patterns of genetic diversity distribution (Wen et al., 2004). In addition to the estimation of forensic characterization of autosomal STRs in northern-Han, we evaluated three different population comparisons to gain a comprehensive genetic overview of the northern-Han Chinese population and nationwide and worldwide reference populations on the basis of the genetic variations of STRs (23-STRs genotype-based data set among 12 Chinese populations, 20-STRs frequency-based dataset among 53 worldwide populations and 19-STRs frequency-based dataset among 61 nationwide populations). Finally, we also collected the present available mitochondrial and Y-chromosomal genetic variations of Han Chinese populations and merged them with previously published uniparental marker variations, as well as combined whole-genome SNPs of modern and Eurasian ancient peoples, to explore the genetic legacy and phylogenetic relationship between northern Han Chinese and ancient populations (Cui et al., 2010; Li et al., 2010; Li et al., 2011; Zhao et al., 2011; Wang et al., 2012; Cui et al., 2013; Zhao et al., 2014; Dong et al., 2015; Gao et al., 2015; Li et al., 2015; Zhao et al., 2015b; Li et al., 2017; Zhang et al., 2017b; Li et al., 2018).
Pengyu Chen1,2†, Jian Wu1,2†, Li Luo1,2†, Hongyan Gao1,2, Mengge Wang3, Xing Zou3, Yingxiang Li4, Gang Chen4, Haibo Luo2, Limei Yu5, Yanyan Han6, Fuquan Jia7* and Guanglin He3*
Modern East Asians derived from the admixture of aborigines and incoming farmers expanding from Yellow and Yangtze River Basins. Distinct genetic differentiation and subsequent admixture between Northeast Asians and Southeast Asians subsequently evidenced by the mitochondrial DNA, Y-chromosomal variations, and autosomal SNPs. Recently, population geneticists have paid more attention to the genetic polymorphisms and background of southern-Han Chinese and southern native populations. The genetic legacy of northern-Han remains uncharacterized. Thus, we performed this comprehensive population genetic analyses of modern and ancient genetic variations aiming to yield new insight into the formation of modern Han, and the genetic ancestry and phylogenetic relationship of the northern-Han Chinese population. We first genotyped 25 forensic associated markers in 3,089 northern-Han Chinese individuals using the new-generation of the Huaxia Platinum System. And then we performed the first meta-analysis focused on the genetic affinity between Asian Neolithic∼Iron Age ancients and modern northern-Han Chinese by combining mitochondrial variations in 417 ancient individuals from 13 different archeological sites and 812 modern individuals, as well as Y-chromosomal variations in 114 ancient individuals from 12 Neolithic∼Iron Age sites and 2,810 modern subjects. We finally genotyped 643,897 genome-wide nucleotide polymorphisms (SNPs) in 20 Shanxi Han individuals and combined with 1,927 modern humans and 40 Eurasian ancient genomes to explore the genetic structure and admixture of northern-Han Chinese. We addressed genetic legacy, population structure and phylogenetic relationship of northern-Han Chinese via various analyses. Our population genetic results from five different reference datasets indicated that Shanxi Han shares a closer phylogenetic relationship with northern-neighbors and southern ethnically close groups than with Uyghur and Tibetan. Genome-wide variations revealed that modern northern-Han derived their ancestry from Yakut-related population (25.2%) and She-related population (74.8%). Summarily, the genetic mixing that led to the emergence of a Han Chinese ethnicity occurred at a very early period, probably in Neolithic times, and this mixing involved an ancient Tibeto-Burman population and a local pre-Sinitic population, which may have been linguistically Altaic.
Introduction
Han Chinese, with a total population size circa 1.4 billion, is the world’s largest ethnic group and dominant ethnicity in China and Singapore. The origin of the Han Chinese population, genetic relationship with adjacent groups and past migratory pattern and admixture history have gained considerable attention from scientists working in the anthropology, linguistics, history, population and forensic genetics (Zhao et al., 2011; Gao et al., 2015; Zhao et al., 2015b; Li et al., 2017; Nothnagel et al., 2017; Zhang et al., 2017b; Chiang et al., 2018). Archaeological and anthropological evidence showed that human occupation in East Asia has experienced archaic hominin extinction, genetic introgression between early anatomically modern human and Denisovan or Neanderthals, the transformation from hunting–gathering to agriculture, massive admixture and migratory history with ethnolinguistically diverse populations in the past 50–100 thousand years (Nielsen et al., 2017). Expansions of the maternally-inherited mitochondrial DNA (mtDNA) and paternally-inherited Y-chromosome haplogroup lineages indicated that ethnically different East Asians derived from southeastern groups and experienced south-to-north migrations driven by a variety of evolutionary mechanisms (Su et al., 1999; Yao et al., 2002). Besides, social practices, including subsistence strategies, residence patterns, and agricultural expansion, play an indispensable role in shaping the patterns of Chinese populations (Nielsen et al., 2017). Ancient mitochondrial and Y-chromosomal DNA studies in East Asian Neolithic∼Iron Age populations have drastically increased in past decades (Cui et al., 2010; Li et al., 2010; Li et al., 2011; Zhao et al., 2011; Wang et al., 2012; Cui et al., 2013; Zhao et al., 2014; Dong et al., 2015; Gao et al., 2015; Li et al., 2015; Zhao et al., 2015b; Li et al., 2017; Zhang et al., 2017b; Li et al., 2018), however, how the peopling and settlement history of Neolithic populations influence the origin, expansion, and migration of the Han Chinese population is still unclear.
Physical anthropological investigation of somatometric and nonmetric features revealed that a significant difference exists between northern-Han Chinese and southern-Han Chinese (Sanchez-Mazas et al., 2011). Subsequently, Chu et al. (1998) found genetic evidence to support the distinction between southern and northern populations. Phylogeographic or genetic differentiation between northern-Han and southern-Han have been also evidenced by Yao et al. (2002) using mitochondrial DNA, Wen et al. (2004) using combined testing Y-chromosome and mitochondrial DNA variations, and Chen et al. (2009) and Xu et al. (2009) using high-density genotype data. Our previous study has investigated the genetic polymorphisms, forensic features and genetic relationship of currently widely-used autosomal short tandem repeats (STRs) in the southern-Han Chinese residing in the Pearl River Delta (He et al., 2018d). Thus, reconstructing the forensic reference database, estimating the forensic allele frequency and parameters and dissecting the genetic relationship of this genetically diverse northern-Han Chinese population are very necessary and urgent.
STR, also called as microsatellite, is one of the extraordinary mutated genetic markers, is widely existed human autosomal, X-chromosome and Y-chromosome genomes (Ge et al., 2014). This length polymorphism marker is generated by the slippage synthesis of simple sequence (2–8bp) (Schlotterer and Tautz, 1992). STRs located on the no-recombining region of Y-chromosome are the best candidates for forensic pedigree searches and identifying the perpetrator in the sexual crime or rape cases, and X-chromosomal STRs are best suitable for applications in the deficiency and incestuous cases (He et al., 2017d; Chen et al., 2018). Autosomal STR genotyping is the gold standard in the routine forensic cases. Nowadays, all organizations or countries optimized their accepted STR panels to improve the international collaboration, such as the expanded CODIS core loci, extended European standard set (ESS-extended). Huaxia Platinum System (Thermo Fisher Scientific) was integrated all twenty expanded CODIS core loci, additional STRs included in the Chinese National Database and two gender determination loci (He et al., 2018b). Single nucleotide polymorphisms (SNPs), with the number over 84.7 million in the human genome, are the best candidate to explore the detailed processes of human origin, migration, evolution, adaptation and admixture (Genomes Project et al., 2015).
Although Y-chromosomal and X-chromosomal variations of the northern-Han Chinese population have been investigated and reported (He et al., 2017d; Chen et al., 2018). Autosomal STR allele distribution of this new-generation of the Huaxia Platinum Amplification System with regard to forensic statistical features has not previously been investigated. Besides, the population genetic structure and admixture history of northern-Han via high-density genetic markers are unclear. Thus, we genotyped and analyzed 23 autosomal STRs in 3,089 unrelated Han Chinese individuals and 643,897 SNPs in 20 Hans residing in Shanxi Province. Shanxi Province is between 34°34′-40°44′ north latitude and 110°14′-114°33′ east longitude, which stretches about a total area of 156,700 km2 from the Yellow River in the west and south to the Taihang Mountain in the east and the Great Wall in the north. This area is bounded by the Shaanxi, Inner Mongolia, Hebei, Henan, and other Provinces. Archaeological, anthropological and genetic evidence from Hengbei site consistently considered that Han Chinese is originated from Shanxi and neighboring regions, also called the Central Plain (Zhao et al., 2015b). And then Han Chinese population migrated southward with the Han-associated culture (Demic diffusion) and admixed with southern Chinese natives and formed the current patterns of genetic diversity distribution (Wen et al., 2004). In addition to the estimation of forensic characterization of autosomal STRs in northern-Han, we evaluated three different population comparisons to gain a comprehensive genetic overview of the northern-Han Chinese population and nationwide and worldwide reference populations on the basis of the genetic variations of STRs (23-STRs genotype-based data set among 12 Chinese populations, 20-STRs frequency-based dataset among 53 worldwide populations and 19-STRs frequency-based dataset among 61 nationwide populations). Finally, we also collected the present available mitochondrial and Y-chromosomal genetic variations of Han Chinese populations and merged them with previously published uniparental marker variations, as well as combined whole-genome SNPs of modern and Eurasian ancient peoples, to explore the genetic legacy and phylogenetic relationship between northern Han Chinese and ancient populations (Cui et al., 2010; Li et al., 2010; Li et al., 2011; Zhao et al., 2011; Wang et al., 2012; Cui et al., 2013; Zhao et al., 2014; Dong et al., 2015; Gao et al., 2015; Li et al., 2015; Zhao et al., 2015b; Li et al., 2017; Zhang et al., 2017b; Li et al., 2018).