|
|
Post by Admin on Feb 12, 2016 5:51:16 GMT
 Zoroastrianism was founded by the Prophet Zoroaster in ancient Iran approximately 3,500 years ago and those who are descended from the Indo-Aryans such as "Yazidis" retained or re-borrowed some practices and concepts from Zoroastrianism from ancient times. The Yezidis are racially similar to Kurds whose R1a frequency is around 11-12% and the name "Yezidi" comes from the ancient city of Yazd in Iran. 11.5% of male Ashkenazim were found to belong to R1a1a, the dominant Indo-European haplogroup in Eastern Europe and Russia, and it's plausible that the Jews and Yazidis share a common Indo-Aryan ancestor, which explains why Judaism borrowed heavily from Zoroastrianism.  Only a few genetic studies have been carried out on Kurdish groups. Previous genetic studies of classical markers (Cavalli-Sforza/Menozzi/ Piazza 1994) indicated a genetic proximity of Kurds to other Middle Eastern populations. Comas et al. (2000) found close European affinities for Kurdish matrilineal lineages while studying the Kurds living in Georgia. Richards et al. (2000), analysing the same genetic markers among Kurds from Eastern Turkey, found that some lineages in Kurdish data set possibly originated in Europe and were associated with back- migrations from Europe to the Near East. Wells et al. (2001) who inves- tigated the distribution of male (Y) chromosome markers in a group of Kurmanji Kurds living in Turkmenistan, has not made any specific in- ferences about the history of this group. Nebel et al. (2001) while studying patrilineal lineages among different groups from the Middle East, found close affinities for the Kurdish sample from northern Iraq to other Middle Eastern groups. Quintana-Murci et al. (2004) studied 20 Kurds from Western Iran and 32 of them from Turkmenistan, among others from Iran, Pakistan, and Central Asia, but did not come to any specific conclusion. Nasidze et al. (2005) studied Kurdish groups in Georgia and came to conclusion that during the migration into the Caucasus they experienced a bottleneck effect, and since that time have not undergone detectable admixture with the Georgians.  A Yazidi refugee child in Zakho, Iraq Hence, the Yezidi community has been strongly isolated from other ethnic and religious groups during at least 7-8 centuries. In the frame- work of this paper, we attempted to elucidate the following points re- lating to the genetic history of this ethnic group: (a) Is it possible to get from the existing data any serious evidence on the common origin of the Yezidis and the Kurds? (b) To what extent long-term reproductive isolation has reflected on the genetic proximity between the Kurds from North Iraq and the Yezidis living in Armenia? (c) Are there any specific features in male lineages for the Yezidi population living in Armenia? DNA samples were collected during 2000 and 2001 from 202 Yezidi males, unrelated at the paternal-grandparental level in six mountainous villages located in central part of Armenia. Comparative data on 95 Kurds were taken from the studies of this community in Northern Iraq (Nebel et al. 2001; Birkman et al. 1999). All samples were screened for 18 genetic markers on male sex chromosome.  The results demonstrate that the Yezidis living in Armenia significantly differ from the Kurds. In the same time, both groups display strong evidence on their Mediterranean origin, bearing in their gene pool the genetic signature of Neolithic farmers (Quinta-Murci et al. 2001): 40,0% of Kurdish lineages belong to this major genetic group, whereas the corresponding figure in the Yezidis is 41,6%. The incidence of the next frequent genetic group displays notable difference between two groups―16,8% in Kurds and 28,2% in Yezidis living in Armenia. The two datasets share only eight lineages out of 94 identified for the pooled sample of the two populations. Among these only two are modal, defined as that occurring in any population at a frequency greater than or equal to 10%. The Kurdish and Yezidi modal lineage is shared with neighbouring or Middle Eastern populations, and also is one of the modal lineages in Turkey and Azerbaijan Republic, and in some geographical groups of Armenians (Weale et al. 2001). In contrast, another Yezidi modal lineage is specific to this population, as its frequencies in other neighbouring ethic groups from the South Caucasus and Middle East are extremely low or even absent (Nebel et al. 2001; Weale at al. 2001; Nebel et al. 2000).  Meanwhile, this lineage is encountered in Kurds at a very small frequency. It is interesting that the major group to which this modal lineage belongs, has just two sources (founders) in the Yezidis, which may reflect that only two clans took part in further spread of this lineage. Genetically, the Yezidi community is significantly less variable than the Kurds. There are at least three reasons causing this difference. Firstly, it could be created at the initial stages of Yezidis’ religious community development due to limited number of people involved in this group and, as a consequence of cultural isolation, rather high rate of inbreeding. Secondly, it might be originated in the course of numerous migrations from Iraq to Turkey and, further, to Armenia. It might be suggested that most members of genetically related families (clans) have migrated from one region to another. The third reason causing de- crease of variability is that the Yezidi community in Armenia is religiously and culturally isolated with only small genetic flow from the Yezidi community living in Georgia―both having been, in fact, parts of the same group of migrants to Transcaucasia. Therefore, smaller genetic diversity of patrilineal lineages in Yezidis could be considered as an inherent feature of the population caused by cultural traditions and the historical destiny of the population. In general, these peculiarities result in significant difference between the two populations considered. Yepiskoposian, Levon et al.. “Genetic Affinity Between the Armenian Yezidis and the Iraqi Kurds”. Iran & the Caucasus 14.1 (2010): 37–42. |
|
|
|
Post by Admin on Mar 1, 2016 1:18:25 GMT
 Figure 1: Genetic structure of ancient Europe. Ancient genomes from Eurasia have revealed three ancestral populations that contributed to contemporary Europeans in varying degrees1. Mesolithic individuals, sampled from Spain all the way to Hungary1, 2, 3, belong to a relatively homogenous group, termed western hunter-gatherers (WHG). The expansion of early farmers (EF) out of the Levant during the Neolithic transition led to major changes in the European gene pool, with almost complete replacement in the south and increased mixing with local WHG further north1, 2, 3, 4, 5. Finally, a later wave originating with the Early Bronze Age Yamnaya from the Pontic steppe, carrying partial ancestry from ancient North Eurasians (ANE) and ancestry from a second, undetermined source, arrived from the east, profoundly changing populations and leaving a cline of admixture in Eastern and Central Europe1, 3, 6. This view, which was initially based on a handful of genomes, was recently confirmed by extensive surveys of Eurasian samples from the Holocene5, 7. Here, we extend our view of the genetic makeup of early Europeans by both looking further back in time and sampling from the crossroads between the European and Asian continents. We sequenced a Late Upper Palaeolithic (‘Satsurblia’ from Satsurblia cave, 1.4-fold coverage) and a Mesolithic genome (‘Kotias’ from Kotias Klde cave, 15.4-fold) from Western Georgia, at the very eastern boundary of Europe. We term these two individuals Caucasus hunter-gatherers (CHG). To extend our overview of WHG to a time depth similar to the one available for our samples from the Caucasus, we also sequenced a western European Late Upper Palaeolithic genome, ‘Bichon’ (9.5-fold) from Grotte du Bichon, Switzerland. These new genomes, together with already published data, provide us with a much-improved geographic and temporal coverage of genetic diversity across Europe after the Last Glacial Maximum (LGM)8. We show that CHG belong to a new, distinct ancient clade that split from WHG ~45 kya and from Neolithic farmer ancestors ~25 kya. This clade represents the previously undetermined source of ancestry to the Yamnaya, and contributed directly to modern populations from the Caucasus all the way to Central Asia.  Figure 2: The relationship between Caucasus hunter-gatherers (CHG), western hunter-gatherers and early farmers. The geographical proximity of the Southern Caucasus to the Levant begs the question of whether CHG might be related to early Neolithic farmers with Near Eastern heritage. To address this question formally we reconstructed the relationship among WHG, CHG and EF using available high-quality ancient genomes1, 3. We used outgroup f3-statistics14 to compare the three possible topologies, with the correct relationship being characterized by the largest amount of shared drift between the two groups that form a clade with respect to the outgroup (Fig. 2a; Supplementary Note 4). A scenario in which the population ancestral to both CHG and EF split from WHG receives the highest support, implying that CHG and EF form a clade with respect to WHG. We can reject a scenario in which CHG and WHG form a distinct clade with respect to EF. The known admixture of WHG with EF1, 3, 4, 5 implies that some shared drift is found between WHG and EF with respect to CHG, but this is much smaller than the shared drift between CHG and EF. Thus, WHG split first, with CHG and EF separating only at a later stage. A partial genome from a 24,000-year-old individual (MA1) from Mal’ta, Siberia6 had been shown to be divergent from other ancient samples and was shown by Lazaridis et al.1, using f4 statistics, to have more shared alleles with nearly all modern Europeans than with an EF genome. This allowed inference of an ANE component in European ancestry, which was subsequently shown to have an influence in later eastern hunter-gatherers and to have spread into Europe via an incursion of Steppe herders beginning ~4,500 years ago5, 7. Several analyses indicate that CHG genomes are not a subset of this ANE lineage. First, MA1 and CHG plot in distinct regions of the PCA and also have very different profiles in the ADMIXTURE analysis (Fig. 1). Second, when we test if CHG shows any evidence of excess allele sharing with MA1 relative to WHG using tests of the form D(Yoruba, CHG; MA1, WHG) no combinations were significantly positive (Supplementary Table 6). Last, we also tested whether the ancestral component inferred in modern Europeans from MA1 was distinct from any that may have been donated from CHG using tests of the form D(Yoruba, MA1; CHG, modern North European population) (Supplementary Table 7). All northern Europeans showed a significant sharing of alleles with MA1 separate to any they shared with CHG.  Figure 3: Distribution of ROH. Given their geographic origin, it seems likely that CHG and EF are the descendants of early colonists from Africa who stopped south of the Caucasus, in an area stretching south to the Levant and possibly east towards Central and South Asia. WHG, on the other hand, are likely the descendants of a wave that expanded further into Europe. The separation of these populations is one that stretches back before the Holocene, as indicated by local continuity through the Late Palaeolithic/Mesolithic boundary and deep coalescence estimates, which date to around the LGM and earlier. Several analyses show that CHG are distinct from another inferred minor ancestral population, ANE, making them a divergent fourth strand of European ancestry that expands the model of the human colonization of that continent. The separation between CHG and both EF and WHG ended during the Early Bronze Age when a major ancestral component linked to CHG was carried west by migrating herders from the Eurasian Steppe. The foundation group for this seismic change was the Yamnaya, who we estimate to owe half of their ancestry to CHG-linked sources. These sources may be linked to the Maikop culture, which predated the Yamnaya and was located further south, closer to the Southern Caucasus. Through the Yamanya, the CHG ancestral strand contributed to most modern European populations, especially in the northern part of the continent.  Figure 4: The relationship of Caucasus hunter-gatherers to modern populations. Finally, we found that CHG ancestry was also carried east to become a major contributor to the Ancestral North Indian component found in the Indian subcontinent. Exactly when the eastwards movement occurred is unknown, but it likely included migration around the same time as their contribution to the western European gene pool and may be linked with the spread of Indo-European languages. However, earlier movements associated with other developments such as that of cereal farming and herding are also plausible. The discovery of CHG as a fourth ancestral component of the European gene pool underscores the importance of a dense geographical sampling of human palaeogenomes, especially among diverse geographical regions. Its separation from other European ancestral strands ended dramatically with the extensive population, linguistic and technological upheavals of the Early Bronze Age resulting in a wide impact of this ancestral strand on contemporary populations, stretching from the Atlantic to Central and South Asia. Nature Communications 6, Article number: 8912 |
|
|
|
Post by Admin on May 12, 2016 22:45:59 GMT
 The oldest Gaelic literature describes the origins of the Irish people as a series of ancient invasions, and the archaeological record in Ireland, as elsewhere in Europe, exhibits several horizons where major cultural shifts are apparent (1). The two most transformative are the arrival of agriculture (∼3750 BC) followed by the onset of metallurgy (∼2300 BC). The Neolithic package characterized by animal husbandry, cereal crops, ceramics, and timber houses reached the shores of Ireland some 5,000 years after its beginnings in the Near East. The second great wave of change starts with the appearance of copper mines, associated with Bell Beaker pottery, which are quickly followed by Bronze tool-making, weaponry, and gold-working, with distinct Food Vessel pottery succeeding from the earlier beakers (2). This period coincides with the end of the large passage graves of Neolithic Ireland in favor of single burials and smaller wedge tombs. High-throughput sequencing has opened the possibility for genome-wide comparisons of genetic variation in ancient populations, which may be informatively set in the context of extensive modern data (4⇓⇓⇓⇓⇓–10). In Europe, these clearly show population replacement by migrating farmers from southwest Asia at the onset of the Neolithic with some retrenchment of the earlier Mesolithic genome at later stages (5⇓⇓⇓–9, 11, 12). Three longitudinal genome studies have also shown later genome-wide shifts around the beginnings of the Bronze Age in central Europe with substantial introgression originating with the Yamnaya steppe herders (7, 9, 10). However, replacement coupled to archaeological horizons is unlikely to be a universal phenomenon, and whether the islands of Britain and Ireland, residing at the temporal and geographical edges of both the Neolithic and steppe migrations, were subject to successive substantial population influxes remains an open and debated question. For example, a recent survey of archaeological opinion on the origins of agriculture in Ireland showed an even split between adoption and colonization as explanatory processes (13). Recent archaeological literature is also divided on the origins of the insular Bronze Age, with most opinion favoring incursion of only small numbers of technical specialists (1, 2, 14, 15).  Fig. 1. Genetic affinities of ancient Irish individuals. (A and B) Genotypes from 82 ancient samples are projected onto the first two principal components defined by a set of 354,212 SNPs from Eurasian populations in the Human Origins dataset (29) (SI Appendix, Section S9.1 and S10). Model-based approaches allow the decomposition of individuals into coefficients contributed by a set number (K) of ancestral populations. We investigated this by using ADMIXTURE (Version 1.23) (27) with ancient individuals included in the analysis alongside 1,941 modern samples from diverse worldwide populations (8) (SI Appendix, Section S11). Fig. 1C shows the plot of estimated ancestry proportions for each ancient genome at a value of K = 11. These partition similarly to previous analyses (7, 9, 10), with three major ancestral coefficients manifesting in west and central Europe. The first (colored red) forms the near totality of ancestry in hunter–gatherer samples, and admixes with a second (orange) component found at high levels in Neolithic and also modern Near Eastern populations (SI Appendix, Section S11.1). Ballynahatty is similar to other MN samples with a majority orange “early farmer” component but with an elevated level of the red “hunter–gatherer” component compared with Early Neolithic genomes. The ancestry of Yamnaya, Early Bronze Age herders from the Pontic Steppe, is evenly divided between this same red component and the third major European coefficient, colored in green, which has been identified with a Caucasus origin (28). This Caucasus component is encountered subsequently, introduced via Yamnaya, in Late Neolithic and Bronze Age samples in central Europe (9, 10) and features in the three Irish Early Bronze Age samples within profiles similar to continental Bronze Age genomes. Bronze Age Replacement. Prior studies (7, 9, 10) convincingly demonstrate that Central European genomes from the late Neolithic and Early Bronze Age differ from the preceding MN due to a substantial introgression originating with Steppe herders linked to cultures such as the Yamnaya. Accordingly, we used a series of tests to gauge whether the ancestries of the Rathlin Early Bronze Age genomes were subject to this influence. D statistics confirmed that Ballynahatty and other MN individuals form clades with each other to the exclusion of these Irish Bronze Age samples. Specific disruption of continuity between the Irish Neolithic and Bronze Age is clear with significant evidence of both Yamnaya and EHG introgression into Irish Bronze Age samples when placed in a clade with any MN. However, like other European Bronze Age samples, this introgression is incomplete, as they also show significant MN ancestry when placed in a clade with Yamnaya. The highest levels of MN ancestry were observed when either Ballynahatty or Gok2 (Scandinavian) was the sample under study. However, when paired with central European Bronze Age populations, the Rathlin samples show no trace of significant introgression from Ballynahatty, suggesting that earlier Irish populations may not have been a source of their partial MN ancestry. These analyses, taken with the PCA and ADMIXTURE results, indicate that the Irish Bronze Age is composed of a mixture of European MN and introgressing Steppe ancestry (9, 10). To estimate the proportion of Yamnaya to MN ancestry in each Irish Bronze Age sample, we took three approaches. First, from ADMIXTURE analysis (Fig. 1), we examined the green Caucasus ancestry component. We presume an ultimate source of this as the Yamnaya where it features at a proportion of 40% of their total ancestry. In our three Irish Bronze Age samples, it is present at levels between 6–13%, which, when scaled up to include the remaining 60% of Yamnaya ancestry, imply a total of 14–33% Yamnaya ancestry and therefore 67–86% MN in the Irish Bronze Age. Second, for each Bronze Age Irish individual, we calculated the proportion of MN ancestry by using the ratio f4(Mbuti, Ballynahatty; X, Dai)/f4(Mbuti, Ballynahatty; Gok2, Dai), which gave estimates between 72 ± 4% to 74 ± 5%, implying again a substantial Yamnaya remainder. Third, we followed the methods described in Haak et al. (9), which use a collection of outgroup populations, to estimate the mixture proportions of three different sources, Linearbandkeramik (Early Neolithic; 35 ± 6%), Loschbour (WHG; 26 ± 12%), and Yamnaya (39 ± 8%), in the total Irish Bronze Age group. These three approaches give an overlapping estimate of ∼32% Yamnaya ancestry.  Fig. 2. Estimated distribution of ROH for ancient samples, placed in the context of values from modern populations. Haplotype-Based Resolution of Continuity. Haplotype-based approaches are more powerful than those using unlinked genetic loci in identifying fine genetic structure, such as that displayed among Europeans, and are relatively robust to bias from marker ascertainment (35, 36). We ran ChromoPainter in fineSTRUCTURE (Version 2) (35) to decompose each ancient genome into a series of haplotypic chunks, and identified which modern individuals from a diverse set of Eurasian populations (37) shared the same, or most similar, haplotype at each given chunk. We then considered the pattern of chunk donation between each ancient genome and modern populations.  Unsurprisingly, the pattern of haplotypic affinity of Ballynahatty among modern European populations is strongly correlated to that of the earlier Neolithic samples (SI Appendix, Fig. S14.2; r > 0.74, P < 10−7), with southern Mediterranean samples in each analysis showing highest levels of chunk copying. However, some differences are discernable; the Hungarian and Stuttgart Neolithic genomes tend toward higher values in eastern Mediterranean (Sicilian, Italian, and Greek samples; Fig. 3 and SI Appendix, Fig. S14.1), and the Irish Neolithic has highest values in the west (Sardinian and Spanish). A further difference lies in the comparison of each to the affinities shown by the Luxembourg WHG, Loschbour, which shows no correlation in its modern affinities with the earlier continental Neolithics but does show a significant relationship (P = 5.4 × 10−4) with those of Ballynahatty, undoubtedly because of greater WHG admixture in her ancestry.  Fig. 3. Comparison of Irish and Central European ancient genomes for haplotype-based affinity to modern populations. Interpolated heatmaps comparing relative haplotype donations by two Irish (Ballynahatty, Rathlin1) and two Hungarian (NE1, BR2) ancient genomes. The most striking feature of the haplotype sharing by the Irish Bronze Age genome is its high median donation levels to Irish, Scottish, and Welsh populations (Fig. 3). In regression with results from the other ancient genomes, these insular Celtic populations, and to a lesser degree the English, show an excess of sharing with Rathlin1, suggesting some level of local continuity at the edge of Europe persisting over 4,000 y. The Hungarian Bronze Age genome shows more affinity with central European populations. Interestingly, for both Bronze Age genomes, the modern Basque population displays outlying low-affinity scores compared with neighboring western European samples, supporting recent findings that suggest a continuity between the Basques and Iberian Chalcolithic groups (25). Phenotypic Analysis. Ireland is unusual in displaying world maximum frequencies of a number of important genetic variants, particularly those involved in lactase persistence and two recessive diseases: cystic fibrosis and hemochromatosis (38⇓⇓–41, 42, 43). Interestingly, both the high coverage Neolithic and Bronze Age individuals were heterozygous for the hemochromatosis alleles H63D and C282Y, respectively. Modern Irish allele frequencies are 15% and 11% for these variants, with the latter, more penetrant variant responsible for a world maximum of this disease in Ireland (44, 45). Additionally and in accordance with other data suggesting a late spread of the lactase persistence phenotype now prevalent in western Europe (7, 10), the Neolithic Ballynahatty was homozygous for the nonpersistent genotype and Rathlin1 was heterozygous and thus tolerant of drinking raw milk into adulthood. We were able to deduce that Neolithic Ballynahatty had a dark hair shade (99.5% probability), most likely black (86.1% probability), and brown eyes (97.3% probability) (46). Bronze Age Rathlin1 probably had a light hair shade (61.4%) and brown eyes (64.3%). However, each Rathlin genome possessed indication of at least one copy of a haplotype associated with blue eye color in the HERC2/OCA2 region. Lara M. Cassidy, 368–373, doi: 10.1073/pnas.1518445113 |
|
|
|
Post by Admin on May 17, 2016 22:44:41 GMT
 Population relationships of samples. Genome-wide ancient DNA from West Eurasia We assembled genome-wide data from 230 ancient individuals from West Eurasia dated to between 6500 and 300 bc (Fig. 1a, Extended Data Table 1, Supplementary Data Table 1 and Supplementary Information section 1). To obtain this data set, we combined published data from 67 samples from relevant periods and cultures4–6, with 163 samples for which we report new data, of which 83 have, to our knowledge, never previously been analysed (the remaining 80 samples include 67 whose targeted single nucleotide polymorphism (SNP) coverage we tripled from 390,000 (‘390k capture’) to 1,240,000 (‘1240k capture’)7; and 13 with shotgun data for which we generated new data using our targeted enrichment strategy3,8). The 163 samples for which we report new data are drawn from 270 distinct individuals who we screened for evidence of authentic DNA7. We used in-solution hybridization with synthesized oligonucleotide probes to enrich promising libraries for the targeted SNPs (Methods). The targeted sites include nearly all SNPs on the Affymetrix Human Origins and Illumina 610-Quad arrays, 49,711 SNPs on chromosome X, 32,681 SNPs on chromosome Y, and 47,384 SNPs with evidence of functional importance. We merged libraries from the same individual and filtered out samples with low coverage or evidence of contamination to obtain the final set of individuals. The 1240k capture gives access to genome-wide data from ancient samples with small fractions of human DNA and increases efficiency by targeting sites in the human genome that will actually be analysed. The effectiveness of the approach can be seen by comparing our results to the largest previously published ancient DNA study, which used a shotgun sequencing strategy5 Insight into population transformations To learn about the genetic affinities of the archaeological cultures for which genome-wide data are reported for the first time here, we studied either 1,055,209 autosomal SNPs when analysing 230 ancient individuals alone, or 592,169 SNPs when co-analysing them with 2,345 present-day individuals genotyped on the Human Origins array4. We removed 13 samples either as outliers in ancestry relative to others of the same archaeologically determined culture, or first-degree relatives (Supplementary Data Table 1). Our sample of 26 Anatolian Neolithic individuals represents the first genome-wide ancient DNA data from the eastern Mediterranean. Our success at analysing such a large number of samples is due to the fact that in the case of 21 of the successful samples, we obtained DNA from the inner ear region of the petrous bone9, which has been shown to increase the amount of DNA obtained by up to two orders of magnitude relative to teeth3. Principal component (PCA) and ADMIXTURE10 analyses show that the Anatolian Neolithic samples do not resemble any present-day near-Eastern populations but are shifted towards Europe, clustering with early European farmers (EEF) from Germany, Hungary and Spain7 (Fig. 1b and Extended Data Fig. 2). Further evidence that the Anatolian Neolithic and EEF were related comes from the high frequency (47%; n= 15) of Y-chromosome haplogroup G2a typical of ancient EEF samples7 (Supplementary Data Table 1), and the low fixa- tion index (FST; 0.005 – 0.016) between Neolithic Anatolians and EEF (Supplementary Data Table 2). These results support the hypothesis7 of a common ancestral population of EEF before their dispersal along distinct inland/central European and coastal/Mediterranean routes. The EEF are slightly more shifted to Europe in the PCA than are the Anatolian Neolithic (Fig. 1b) and have significantly more admixture from Western hunter-gatherers (WHG), as shown by f4-statistics (|Z| > 6 standard errors from 0) and negative f3-statistics (|Z| > 4)11 (Extended Data Table 2). We estimate that the EEF have 7–11% more WHG admixture than their Anatolian relatives (Extended Data Fig. 2, Supplementary Information section 2).  Genome-wide scan for selection. The Iberian Chalcolithic individuals from El Mirador cave are genet- ically similar to the Middle Neolithic Iberians who preceded them (Fig. 1b and Extended Data Fig. 2), and have more WHG ancestry than their Early Neolithic predecessors7 (|Z|> 10) (Extended Data Table 2). However, they do not have a significantly different proportion of WHG ancestry (we estimate 23–28%) than the Middle Neolithic Iberians (Extended Data Fig. 2). Chalcolithic Iberians have no evi- dence of steppe ancestry (Fig. 1b and Extended Data Fig. 2), in contrast to central Europeans of the same period5,7. Thus, the steppe-related ancestry that is ubiquitous across present-day Europe4,7 arrived in Iberia later than in central Europe (Supplementary Information section 2). To understand population transformations in the Eurasian steppe, we analysed a time transect of 37 samples from the Samara region spanning ~5600–1500 bc and including the Eastern hunter-gath- erer (EHG), Eneolithic, Yamnaya, Poltavka, Potapovka and Srubnaya cultures. Admixture between populations of Near Eastern ancestry and the EHG7 began as early as the Eneolithic (5200–4000 bc), with some individuals resembling EHG and some resembling Yamnaya (Fig. 1b and Extended Data Fig. 2).  Allele frequencies for five genome-wide significant signals of selection. The Yamnaya from Samara and Kalmykia, the Afanasievo people from the Altai (3300–3000 bc), and the Poltavka Middle Bronze Age (2900–2200 bc) population that followed the Yamnaya in Samara are all genetically homogeneous, forming a tight ‘Bronze Age steppe’ cluster in PCA (Fig. 1b), sharing predom- inantly R1b Y chromosomes5,7 (Supplementary Data Table 1), and having 48–58% ancestry from an Armenian-like Near Eastern source (Extended Data Table 2) without additional Anatolian Neolithic or EEF ancestry7 (Extended Data Fig. 2). After the Poltavka period, popula- tion change occurred in Samara: the Late Bronze Age Srubnaya have ~17% Anatolian Neolithic or EEF ancestry (Extended Data Fig. 2). Previous work documented that such ancestry appeared east of the Urals beginning at least by the time of the Sintashta culture, and suggested that it reflected an eastward migration from the Corded Ware peoples of central Europe5. However, the fact that the Srubnaya also had such ancestry indicates that the Anatolian Neolithic or EEF ancestry could have come into the steppe from a more eastern source. Further evidence that migrations originating as far west as central Europe may not have had an important impact on the Late Bronze Age steppe comes from the fact that the Srubnaya possess exclusively (n= 6) R1a Y chromosomes (Supplementary Data Table 1), and four of them (and one Poltavka male) belonged to haplogroup R1a-Z93, which is common in central/ south Asians12, very rare in present-day Europeans, and absent in all ancient central Europeans studied to date. The strongest signal of selection is at the SNP (rs4988235) responsible for lactase persistence in Europe15,16. Our data (Fig. 3) strengthens previous reports that an appreciable frequency of lactase persistence in Europe only dates to the last 4,000 years3,5,17. The allele’s earliest appearance in the dataset is in a central European Bell Beaker sample (individual I0112) dated to between 2450 and 2140 bc. Two other independent signals related to diet are located on chromosome 11 near FADS1 and DHCR7. FADS1 and FADS2 are involved in fatty acid metabolism, and variation at this locus is associated with plasma lipid and fatty acid concentration18. The selected allele of the most significant SNP (rs174546) is associated with decreased triglyceride levels18. This locus has experienced independent selection in non-Eu- ropean populations13,19,20 and is likely to be a critical component of adaptation to different diets. Variants at DHCR7 and NADSYN1 are associated with circulating vitamin D levels21 and the most associ- ated SNP in our analysis, rs7940244, is highly differentiated across closely related northern European populations22,23, suggesting selection related to variation in dietary or environmental sources of vitamin D. Two signals have a potential link to coeliac disease. One occurs at the ergothioneine transporter SLC22A4 that is hypothesized to have experienced a selective sweep to protect against ergothioneine deficiency in agricultural diets24. Common variants at this locus are associated with increased risk for ulcerative colitis, coeliac disease, and irritable bowel disease and may have hitchhiked to high frequency as a result of this sweep24–26. However, the specific variant (rs1050152, L503F) that was thought to be the target did not reach high frequency until relatively recently (Extended Data Fig. 4). The signal at ATXN2/SH2B3—also associated with coeliac disease25—shows a similar pattern (Extended Data Fig. 4). The second strongest signal in our analysis is at the derived allele of rs16891982 in SLC45A2, which contributes to light skin pigmentation and is almost fixed in present-day Europeans but occurred at much lower frequency in ancient populations. In contrast, the derived allele of SLC24A5 that is the other major determinant of light skin pigmentation in modern Europe (and that is not significant in the genome-wide scan for selection) appears fixed in the Anatolian Neolithic, suggesting that its rapid increase in frequency to around 0.9 in Early Neolithic Europe was mostly due to migration (Extended Data Fig. 4). Another pigmentation signal is at GRM5, where SNPs are associated with pigmentation possibly through a regulatory effect on nearby TYR27. 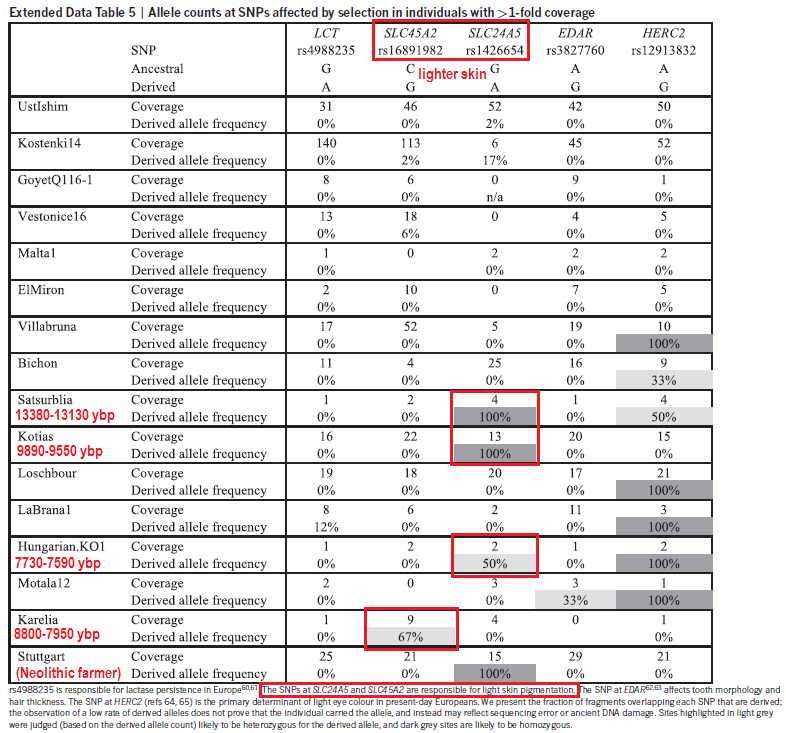 We also find evidence of selection for the derived allele of rs12913832 at HERC2/OCA2, which is at 100% frequency in the European hunter- gatherers we analysed, and is the primary determinant of light eye colour in present-day Europeans28,29. In contrast to the other loci, the range of frequencies in modern populations is within that of ancient populations (Fig. 3). The frequency increases with higher latitude, suggesting a complex pattern of environmental selection. The TLR1–TLR6–TLR10 gene cluster is a known target of selection in Europe, possibly related to resistance to leprosy, tuberculosis or other mycobacteria30–32. There is also a strong signal of selection at the major histocompatibility complex (MHC) on chromosome 6. The strongest signal is at rs2269424 near the genes PPT2 and EGFL8, but there are at least six other apparently independent signals in the MHC (Extended Data Fig. 3); and the entire region is significantly more associated than the genome-wide average (residual inflation of 2.07 in the region on chromosome 6 between 29–34 Mb after genome-wide genomic control correction). This could be the result of multiple sweeps, balancing selection, or increased drift as a result of background selection reducing effective population size in this gene-rich region. Nature 528, 499–503 (24 December 2015) |
|
|
|
Post by Admin on Jun 18, 2016 22:50:49 GMT
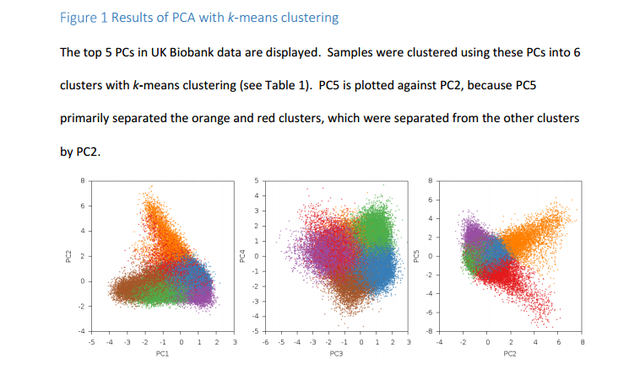 Analyses with ancient Eurasians show that populations in the northern UK have higher levels of Steppe ancestry, and that UK population structure cannot be explained as a simple mixture of Celts and Saxons. A scan for unusual population differentiation along top PCs identified a genome-wide significant signal of selection at the coding variant rs601338 in FUT2 (p=9.16×10-9) We consistently obtained significantly positive f4 statistics, implying that both the modern Celtic samples and the ancient Saxon samples have more Steppe ancestry than the modern Anglo-Saxon samples from southern and eastern England. This indicates that southern and eastern England is not exclusively a genetic mix of Celts and Saxons. 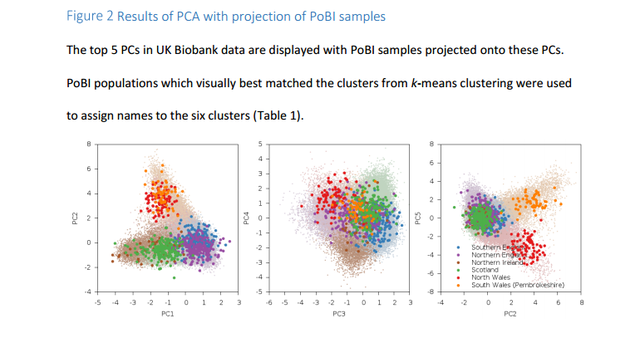 Southeastern England is genetically very homogeneous. If the people there were a mix of ancient Celts and Saxons you'd expect them to be intermediate between modern Celts (who should have more Celtic ancestry than the modern English) and ancient Saxons (who should have more Saxon ancestry than the modern English). But, it seems that the English have less steppe ancestry than both modern Celts and ancient Saxons, so they're not really intermediate. The establishment of Viking settlements in the 9th century in such places as East Anglia, Yorkshir and Dublin could also have played a role in the selection of sexual partners and women may have preferred tall and blonde men of Scandinavian decent or vise versa. The ancient Saxons had more Yamna steppe ancestry than modern Anglo-Saxons and Viking migrations to Britan may have diluted modern Anglo-Saxons' Yamna steppe ancestry. 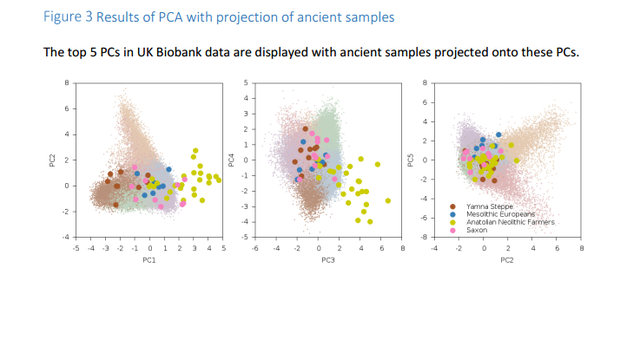 In addition to the impact of ancient Eurasian populations, we know that the genetics of the UK has been strongly impacted by Anglo-Saxon migrations since the Iron Age24, with the Angles arriving in eastern England and the Saxons in southern England. The Anglo-Saxons interbred with the native Celts, which explains much of the genetic landscape in the UK. We analyzed a variety of samples from Celtic (Scotland and Wales) and Anglo-Saxon (southern and eastern England) populations from modern Britain in conjunction with the PoBI samples20 and 10 ancient Saxon samples from eastern England24 in order to assess the relative amounts of Steppe ancestry. We computed f4 statistics27 of the form f4 (Steppe, Neolithic Farmer; Pop1, Pop2), where Steppe and Neolithic Farmer populations are from ref. 21,22, Pop1 is either a modern Celtic or ancient Saxon population and Pop2 is a modern Anglo-Saxon population (Table 2, Supplementary Table 2).  This statistic is sensitive to Steppe ancestry with positive values indicating more Steppe ancestry in Pop1 than Pop2. We consistently obtained significantly positive f4 statistics, implying that both the modern Celtic samples and the ancient Saxon samples have more Steppe ancestry than the modern Anglo-Saxon samples from southern and eastern England. This indicates that southern and eastern England is not exclusively a genetic mix of Celts and Saxons. There are a variety of possible explanations, but one is that the present genetic structure of Britain, while subtle, is quite old, and that southern England in Roman times already had less Steppe ancestry than Wales and Scotland.  doi: dx.doi.org/10.1101/055855 |
|

