|
|
Post by Admin on May 17, 2023 20:09:33 GMT
DNA study shows migration patterns of ancient Mexican civilizations much more complex than expected 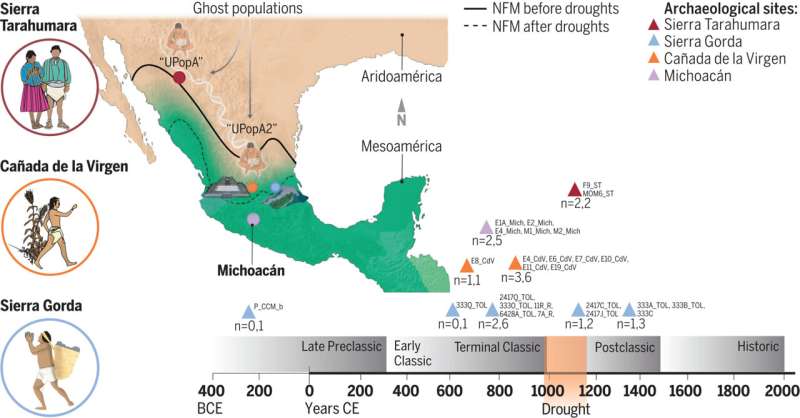 An international team of biologists, geneticists, anthropologists and biochemists has found, through genetic analysis, that the migration patterns of ancient Mexican civilizations were much more complex than previously thought. In their study, reported in the journal Science, the group generated genomic and mitochondrial DNA data to test theories surrounding the migration of ancient peoples in Mexico. Bastien Llamas and Xavier Roca-Rada with the University of Adelaide have published a Perspectives piece in the same journal issue outlining the ethical approach used by the research team to learn more about ancient Mexico. Prior research, based mostly on archaeological evidence, has suggested that drought-driven migration of ancient people from Mexico's north to the south occurred many times in the years before Europeans arrived. The northern region, called Aridoamerica, was dry and mostly desert. The people living there at the time survived as hunter-gatherers. Farther south was Mesoamerica, where early people survived by farming. Prior research has shown that there were several long-term droughts in Aridoamerica, leading people to move south. But now it appears that these conclusions were in error. Instead of relying on archaeological evidence, the team on this new effort looked at the DNA of people living there to see if they were migrating. To learn more about the history of the people living in what is now Mexico, the researchers analyzed DNA samples going back approximately 2,300 years. In all, they were able to study 27 samples obtained from eight archaeological sites from people who lived in regions of what is now Mexico. The researchers could see that the expected migrations had not occurred. They point out, for example, that despite droughts, sometimes decades long, people living in Sierra Gorda did not leave. The team found none of their DNA in people living farther south. The research team was not able to explain why the northerners had not migrated south when conditions grew dry, but suggest it might have been related to cinnabar commerce. The mineral was easily found in the north, and was sacred to people in the south—thus it seems trade was likely. Regardless of the reasons, the research team suggests migration patterns in early Mexico were far more complex than previously thought. More information: Viridiana Villa-Islas et al, Demographic history and genetic structure in pre-Hispanic Central Mexico, Science (2023). DOI: 10.1126/science.add6142 Bastien Llamas et al, Paleogenomic study of the Mexican past, Science (2023). DOI: 10.1126/science.adh7902 Journal information: Science www.science.org/doi/10.1126/science.add6142 |
|
|
|
Post by Admin on Jul 13, 2023 7:26:02 GMT
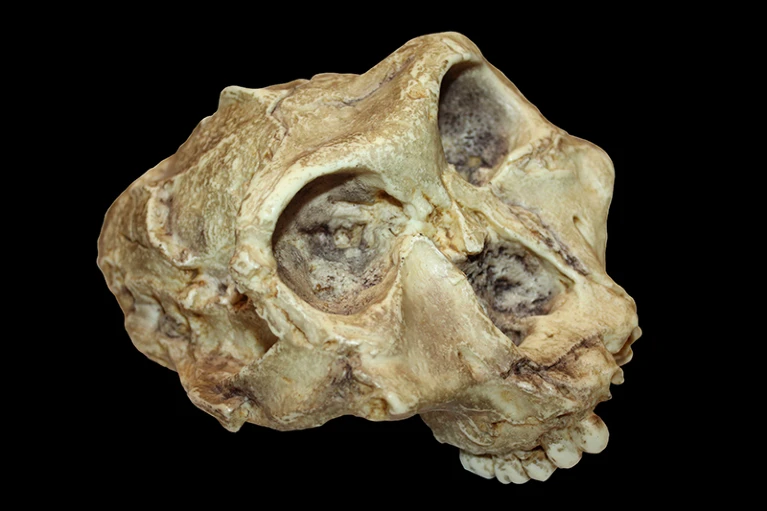 Hominins — humans and their ancient relatives — emerged in Africa some seven million years ago. Now researchers have gleaned genetic information from an African hominin that lived two million years ago, the oldest such data yet recovered. The protein sequences, described in a preprint posted on the bioRxiv server on 3 July1, come from several Paranthropus robustus tooth fossils found in a South African cave. These genetic data are the oldest that have been collected from any hominin, pushing back the genetic record to times and places previously unthinkable, scientists say. “It’s an amazing result,” says Katerina Douka, an archaeological scientist at the University of Vienna. At that age, the remains have “almost turned into stone”, she adds. It is unclear whether the few sequences that can be recovered from very old fossils can help to untangle evolutionary relationships that scientists have debated over for decades. “Nobody really knows yet how useful this will be,” says Beatrice Demarchi, a biomolecular archaeologist at the University of Turin, Italy. Preserved proteins Last year, researchers obtained genetic sequences from two-million-year-old samples of Greenland permafrost2, setting the record for the oldest discovery of preserved ancient DNA. But DNA degrades quicker in warmer climates. Researchers went to heroic efforts to sequence a sliver of the oldest hominin DNA on record: a 400,000-year-old Neanderthal genome, which was found in an underground pit in Spain3. Proteins tend to be more resilient than DNA, allowing researchers to push the molecular record further back in time. In 2016, Demarchi’s team obtained protein sequences from ostrich (Struthionidae) eggshells in Tanzania that were up to 3.8 million years old4. Several years later, a team co-led by Enrico Cappellini, a protein chemist at the University of Copenhagen (KU), sequenced tooth proteins from roughly 800,000-year-old remains belonging to a species called Homo antecessor in Spain, as well as more limited sequences from 1.8-million-year-old Homo erectus fossils from Georgia5. In the 2023 study, a team led by Cappellini and his colleagues at KU — protein scientist Claire Koenig, molecular biologist Ioannis Pastramanis and molecular biologist Palesa Madupe — sampled four P. robustus teeth from Swartkrans cave, 40 kilometres northwest of Johannesburg. Researchers have long debated how these stout-bodied hominins are related to other ancient human species. The researchers used a technique called mass spectrometry to analyse hundreds of amino acids in each sample’s enamel, the mineral outer layer of teeth. One protein they found, called amelogenin-Y, is produced by a gene on the Y chromosome. Its presence in two of the samples allowed the researchers to conclude that the teeth belonged to males. One of these had previously been attributed to a female on the basis of its small size. The other two teeth lacked signs of amelogenin-Y and contained the X-chromosome version of the protein, leading the authors to deduce that the specimens were probably from females. Around 400 of the same amino acids were sequenced in all four samples. This allowed the researchers to build a simple evolutionary tree confirming that Homo sapiens, Neanderthals and hominins found in Siberia called Denisovans that lived in the past 200,000 years are all more closely related to one another than they are to the two-million-year-old Paranthropus. Any other relationship would have been a big surprise, says Douka. In one enamel protein, the researchers found sequence differences between the Paranthropus remains, potentially reflecting variability within the species. ‘Potentially transformative’ Building an evolutionary tree from genetic data of such old remains “can be considered a potentially transformative breakthrough for palaeoanthropology”, Cappellini and his colleagues write in the preprint. They add that ancient-protein studies could improve understanding of where creatures such as Australopithecus afarensis — of which there are many fossil fragments and the more complete specimen known as Lucy — sit in the hominin family tree. Other scientists say that the jury is still out on whether ancient proteins will help bring consensus to the picture of hominin evolution, which is currently built largely from the shapes of bones. There is limited variability in enamel proteins, so the 425 amino acids Cappellini’s team used to construct the family tree seem less informative than the first Neanderthal sequences researchers sequenced in 1997; those included around 360 base pairs of mitochondrial DNA6, which carries lots of variation, notes Pontus Skoglund, a palaeogeneticist at the Francis Crick Institute in London. Bone shape is probably still a more reliable way than ancient proteins as a way to untangle relationships. “There is thus still a long way to go in evolutionary ancient proteomics.” Demarchi agrees, but she’s also excited at the possibility of determining the sex of fragmentary fossils, particularly those of animals. It’s not uncommon for sex-based differences in size to be mistakenly attributed to species differences, and vice versa: P. robustus fossils were initially attributed to males of another, smaller southern African hominin. As ancient proteomics expands, researchers say that it will also be key to balance the benefits against the costs of destructive sampling. Skoglund’s lab is working on non-destructive methods to screen fossils for their protein content — sparing samples that wouldn’t have yielded good data. “It is important that the risk of failure is considered when taking decisions about sampling the fossil record,” he says. www.biorxiv.org/content/10.1101/2023.07.03.547326v1 |
|
|
|
Post by Admin on Aug 1, 2023 2:02:37 GMT
Linguistics and genetics combine to propose a new hybrid theory regarding the origin of the Indo-European languages. For over two centuries, the question of where the Indo-European languages originated has been a hotbed of contention. Two prevailing theories have recently taken center stage in this discussion: the ‘Steppe’ hypothesis, postulating that these languages originated around 6000 years ago in the Pontic-Caspian Steppe, and the ‘Anatolian’ or ‘farming’ hypothesis, which posits an earlier origin connected to early agriculture around 9000 years ago. Previous phylogenetic investigations into the Indo-European languages have yielded contradictory results regarding the age of this language family. This discordance can be attributed to a mix of inaccuracies and inconsistencies in the datasets used in these studies, as well as constraints in the way phylogenetic methodologies have examined ancient languages. To solve these problems, researchers from the Department of Linguistic and Cultural Evolution at the Max Planck Institute for Evolutionary Anthropology assembled an international team of over 80 language specialists to construct a new dataset of core vocabulary from 161 Indo-European languages, including 52 ancient or historical languages. This more comprehensive and balanced sampling, combined with rigorous protocols for coding lexical data, rectified the problems in the datasets used by previous studies. 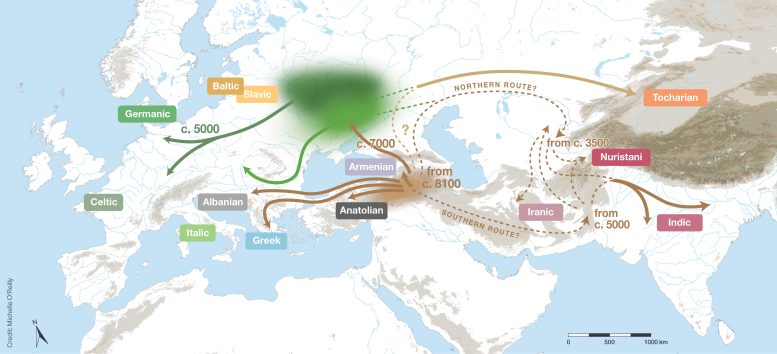 Indo-European estimated to be around 8100 years old The team used recently developed ancestry-enabled Bayesian phylogenetic analysis to test whether ancient written languages, such as Classical Latin and Vedic Sanskrit, were the direct ancestors of modern Romance and Indic languages, respectively. Russell Gray, Head of the Department of Linguistic and Cultural Evolution and senior author of the study, emphasized the care they had taken to ensure that their inferences were robust. “Our chronology is robust across a wide range of alternative phylogenetic models and sensitivity analyses,” he stated. These analyses estimate the Indo-European family to be approximately 8100 years old, with five main branches already split off by around 7000 years ago. These results are not entirely consistent with either the Steppe or the farming hypotheses. The first author of the study, Paul Heggarty, observed that “Recent ancient DNA data suggest that the Anatolian branch of Indo-European did not emerge from the Steppe, but from further south, in or near the northern arc of the Fertile Crescent — as the earliest source of the Indo-European family. Our language family tree topology, and our lineage split dates, point to other early branches that may also have spread directly from there, not through the Steppe.” New insights from genetics and linguistics The authors of the study, therefore, proposed a new hybrid hypothesis for the origin of the Indo-European languages, with an ultimate homeland south of the Caucasus and a subsequent branch northwards onto the Steppe, as a secondary homeland for some branches of Indo-European entering Europe with the later Yamnaya and Corded Ware-associated expansions. “Ancient DNA and language phylogenetics thus combine to suggest that the resolution to the 200-year-old Indo-European enigma lies in a hybrid of the farming and Steppe hypotheses”, remarked Gray. Wolfgang Haak, a Group Leader in the Department of Archaeogenetics at the Max Planck Institute for Evolutionary Anthropology, summarizes the implications of the new study by stating, “Aside from a refined time estimate for the overall language tree, the tree topology and branching order are most critical for the alignment with key archaeological events and shifting ancestry patterns seen in the ancient human genome data. This is a huge step forward from the mutually exclusive, previous scenarios, towards a more plausible model that integrates archaeological, anthropological, and genetic findings.” Reference: “Language trees with sampled ancestors support a hybrid model for the origin of Indo-European languages” by Paul Heggarty, Cormac Anderson, Matthew Scarborough, Benedict King, Remco Bouckaert, Lechosław Jocz, Martin Joachim Kümmel, Thomas Jügel, Britta Irslinger, Roland Pooth, Henrik Liljegren, Richard F. Strand, Geoffrey Haig, Martin Macák, Ronald I. Kim, Erik Anonby, Tijmen Pronk, Oleg Belyaev, Tonya Kim Dewey-Findell, Matthew Boutilier, Cassandra Freiberg, Robert Tegethoff, Matilde Serangeli, Nikos Liosis, Krzysztof Stroński, Kim Schulte, Ganesh Kumar Gupta, Wolfgang Haak, Johannes Krause, Quentin D. Atkinson, Simon J. Greenhill, Denise Kühnert and Russell D. Gray, 28 July 2023, Science. DOI: 10.1126/science.abg0818 |
|
|
|
Post by Admin on Aug 9, 2023 0:14:33 GMT
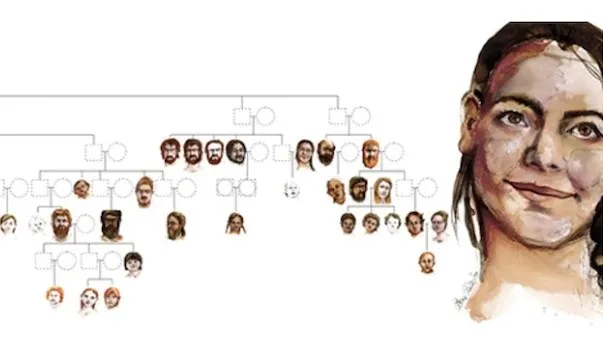 An artist's interpretation of what each individual may have looked like, based on DNA. The dotted squares and circles represent male and female individuals, respectfully, who were not found at the site or whose remains lacked significant DNA. (Image credit: Drawing by Elena Plain; reproduced with the permission of the University of Bordeaux / PACEA) Using ancient DNA, archaeologists in France have pieced together two elaborate Neolithic family trees that span multiple generations, making them the largest ancestral human record ever reconstructed. The family trees are based on a 6,700-year-old funerary site known as Gurgy, which is located in the Paris Basin region of northern France. Researchers excavated the site in the mid-2000s but, due to advancements in obtaining and analyzing ancient DNA data, recently began studying the genomes of 94 of the 128 individuals, which included children and adults, whose remains were recovered from the site, according to a study published July 26 in the journal Nature. Neolithic communities first emerged roughly 12,000 years ago in the Near East, a region that encompasses West Asia, Southeastern Europe and North Africa. During this time period, many human groups transitioned from hunting and gathering to farming. This lifestyle change enabled people to put down roots and settle into communities that spread across generations, leading to the extensive burial plot. "The size of a family tree that huge for that time period" was mind boggling, lead study author Maïté Rivollat, a postdoctoral fellow in the Department of Archaeology at Ghent University in Belgium, told Live Science. "We realized that we could explore social aspects of this community." Related: Largest family tree ever created retraces the history of our species The site was composed of a single graveyard with no monument or grave goods, and many of the bones were "not well preserved and corroded," Rivollat said. Still, "the bones were good enough [to extract] DNA," she said, "and we were able to get DNA from 94 of the individuals."  Researchers discovered that the family's descendants stemmed from a single "founding father." His skeleton was unique, since it was initially buried at an unknown site and was later moved near his kin at Gurgy, according to a statement. (The archaeologists also found the remains of a woman buried next to him but were unable to extract any DNA.) By analyzing the mitochondrial DNA (maternal lineages) and Y-chromosome (paternal lineages) data, as well as each individual's age at death and genetic sex, the researchers constructed two family trees. The first tree connected 64 individuals across seven generations and is the largest to date, and the second contained 12 people from five generations, according to the study. Soon, a "patrilineal pattern" emerged in which generations were linked through the male line of descendants. Researchers also noticed that while the men stayed within the community in which they were born, the women left, according to the statement. "The women who were buried there weren't related and came from somewhere else," Rivollat said. "We also noticed that inbreeding wasn't occurring and think that this system of female movements avoided that from happening." Another interesting aspect of the community was that it lacked half-siblings and that sons and daughters shared the same parents, suggesting that members of this family group weren’t polygamous but rather were monogamous, according to the statement. www.nature.com/articles/s41586-023-06350-8 |
|
|
|
Post by Admin on Aug 9, 2023 23:34:59 GMT
60,000 years of interactions between Central and Eastern Africa documented by major African mitochondrial haplogroup L2  Schematic phylogeny of mtDNA haplogroup L2, based on ML age estimates. Colour scheme corresponding to the probable origin of each clade (WA – Western Africa, CA – Central Africa, EA – Eastern Africa, SA – Southern Africa, EUR – Europe, NE – Near East/Arabian Peninsula), new branch labels proposed in the present study are underlined. Mitochondrial DNA (mtDNA) haplogroup L2 originated in Western Africa but is nowadays spread across the entire continent. L2 movements were previously postulated to be related to the Bantu expansion, but L2 expansions eastwards probably occurred much earlier. By reconstructing the phylogeny of L2 (44 new complete sequences) we provide insights on the complex net of within-African migrations in the last 60 thousand years (ka). Results show that lineages in Southern Africa cluster with Western/Central African lineages at a recent time scale, whereas, eastern lineages seem to be substantially more ancient. Three moments of expansion from a Central African source are associated to L2: (1) one migration at 70-50 ka into Eastern or Southern Africa, (2) postglacial movements (15-10 ka) into Eastern Africa; and (3) the southward Bantu Expansion in the last 5 ka. The complementary population and L0a phylogeography analyses indicate no strong evidence of mtDNA gene flow between eastern and southern populations during the later movement, suggesting low admixture between Eastern African populations and the Bantu migrants. This implies that, at least in the early stages, the Bantu expansion was mainly a demic diffusion with little incorporation of local populations.  Frequency distribution maps for mtDNA haplogroup L2. Maps for L2a (a), L2b (b), L2* (c), L2d (d), L2e (e) and total L2 (f). The map was obtained from the website www.outline-world-map.com. 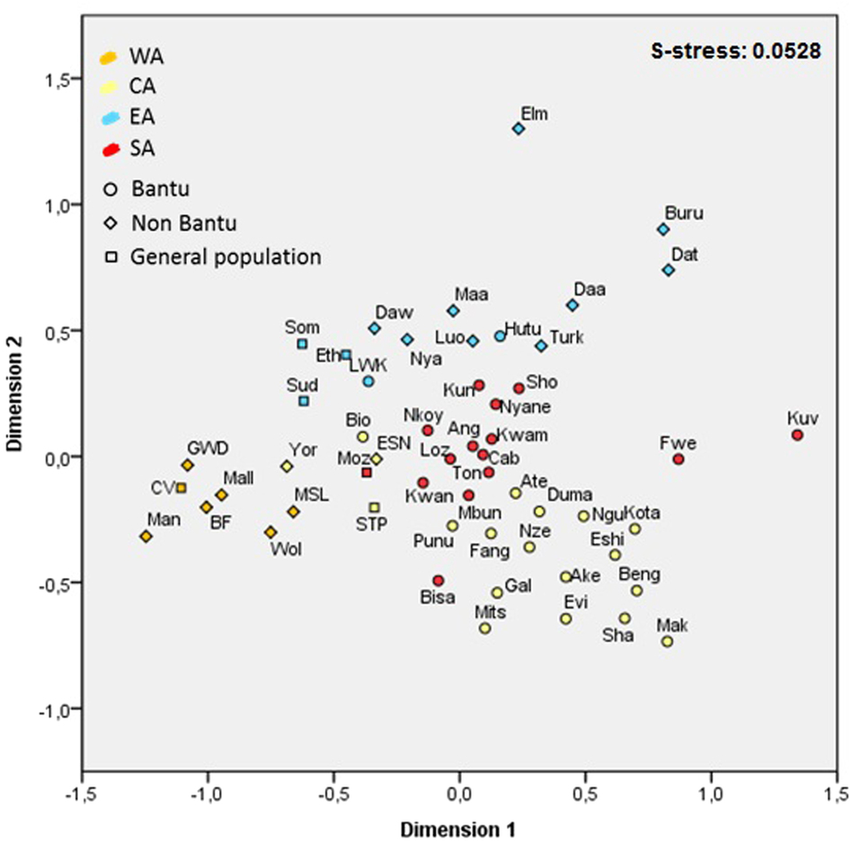 MDS plot based on Slatkin’s linearized FST. Colour code: WA – Western Africa, CA – Central Africa, EA – Eastern Africa, SA – Southern Africa.  Haplogroup composition of sub-Saharan African countries. Population abbreviations: BF – Burkina Faso, CV – Cape Verde, S. Africa – South Africa, S. Leone – Sierra Leone, STP – São Tomé and Príncipe, W. Pygmies – Western Pygmies. The map was obtained from the website www.outline-world-map.com. www.researchgate.net/publication/280496425_60000_years_of_interactions_between_Central_and_Eastern_Africa_documented_by_major_African_mitochondrial_haplogroup_L2 |
|

