|
|
Post by Admin on Mar 25, 2023 11:33:44 GMT
Population genomics unravels the Holocene history of bread wheat and its relatives in Paleogenetics Abstract Deep knowledge of crop biodiversity is essential to improve global food security. Despite bread wheat serving as a keystone crop worldwide, the population history of bread wheat and its wild relatives (a.k.a. wheats) remains elusive. By analyzing whole-genome sequences of 795 wheats, we found that bread wheat originated southwest of the Caspian Sea ∼11,700 years ago and underwent a slow speciation process, lasting ∼3,300 years due to persistent gene flow from wild relatives. Soon after, bread wheat spread across Eurasia and reached Europe, South Asia, and East Asia ∼7,000 to ∼5,000 years ago, shaping a diversified but occasionally convergent adaptive landscape of bread wheat in novel environments. Opposite to cultivated wheat, wild wheat populations have declined by ∼82% in the past ∼2,000 years due to the food choice shift of humans, and likely continue to drop because of the changing climate. These findings will guide future efforts in protecting and utilizing wheat biodiversity to improve global food security. 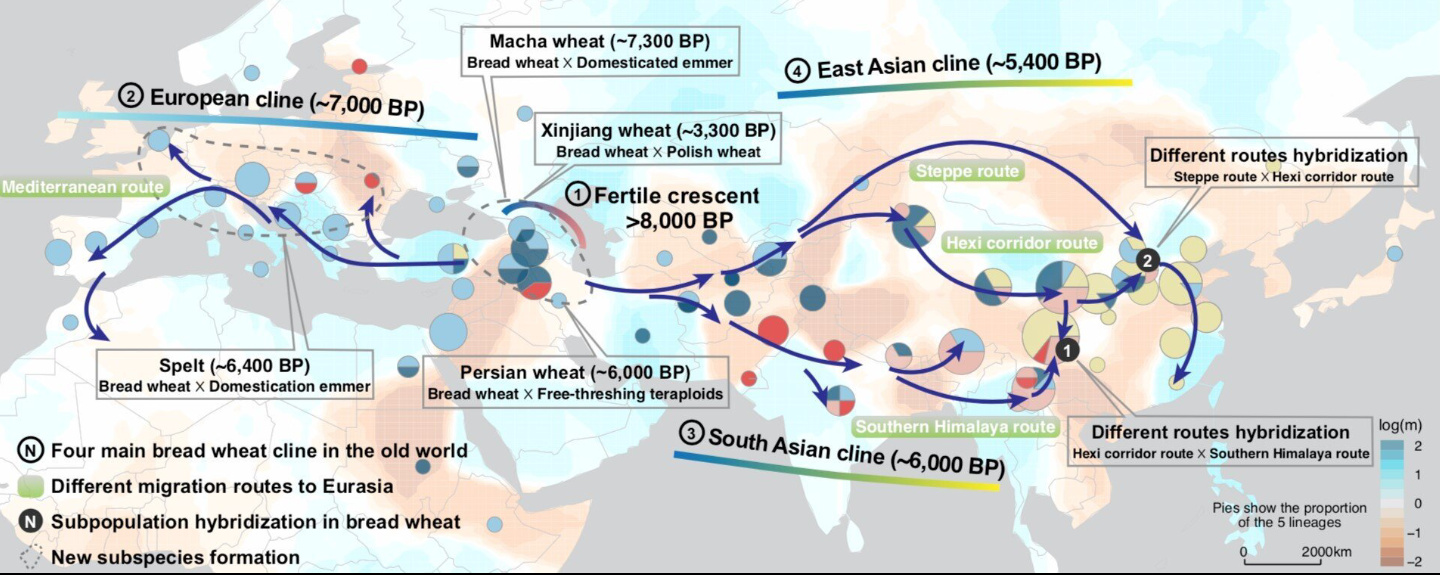 Introduction Climate change and the growing population are putting global food security at risk—the world crop production is projected to be inadequate by 20501. While various adaptive strategies2 and technologies3 of plant breeding have been proposed to address the challenge, many of these opportunities lie in crop biodiversity, which preserves tremendous pre-adapted and beneficial alleles to develop productive, nutritious, stress-resilient, and sustainable crop varieties4. An in-depth understanding of cultivated crops and their wild relatives is central to integrating genetic resources and breeding methods effectively. Bread wheat (Triticum aestivum ssp. aestivum, 2n = 6x = 42, AABBDD) is one of the world’s most important crops, providing ∼20% calories and protein for the human diet5. Meanwhile, bread wheat and its relatives, such as domesticated einkorn (T. monococcum ssp. monococcum, AA) and domesticated emmer (T. turgidum ssp. dicoccum, AABB), were among the first crops bringing forth agriculture and subsequent civilization6. Due to the economic and cultural importance of these ancient crops, the evolutionary history of Triticum and Aegilops species, the two clades giving rise to modern bread wheat through polyploidization7, has been of great interest to both scientists7–9 and the public10,11. Fueled by the landmark bread wheat reference genome12, recent studies have reconstructed the phylogeny of Triticum-Aegilops species13,14, characterized the population structure of modern wheat9,13,15–17, and identified historical gene flow from wild populations to bread wheat13,14,16,18,19. However, the population history of wheats (bread wheat and its wild relatives, or Triticum-Aegilops species) is largely incomplete, particularly the spatiotemporal dynamics of bread wheat emergence and dispersal, together with the genetic and ecological interaction between bread wheat and its wild relatives, remain elusive6,20,21. Here we performed a genus-level sampling of Triticum-Aegilops species and conducted whole-genome sequence analyses to disentangle the deep past of wheats since the rise of agriculture ∼10,000 years ago. The paralleled reconstruction of demographic histories of both cultivated and wild wheats provided the first example of the Holocene evolution of the entire gene pool appertaining to a crop species, insights from which will benefit biodiversity conservation and breeding of many crops. www.biorxiv.org/content/10.1101/2022.04.07.487499v1.full |
|
|
|
Post by Admin on Mar 26, 2023 18:42:36 GMT
Results Genomic data of Triticum-Aegilops populations We collected whole-genome sequencing data of 795 accessions, including 745 accessions from publicly available data set13,17,18, and 50 newly sequenced accessions in this study to complete the sampling of wild relatives of bread wheat. These highly diverse accessions are from 6 species and 25 subspecies in the genera Triticum and Aegilops (Fig. 1), representing a wide range of geographic distribution (73 countries, Supplementary Fig. 1), comprehensive ploidy levels (diploid, tetraploid, and hexaploid) and genome types (AA, BB/SS, AABB, AABBDD, and DD) related to the A, B, and D subgenomes of bread wheat, as well as distinct breeding status (wild progenitors, early domesticates, landraces, and cultivars) (Supplementary Table 1 and 2; for convenience, the common names of subspecies are used in this study). Notably, the collection also well represents the evolutionary trajectory of modern bread wheat7,8,14 (Fig. 1c).  Fig. 1 The representative collection of wheats in this study. a, Common name, spike morphology, ploidy level, genome type, and breeding status of wheat accessions. b, Relationship of wheat accessions in the AB lineage illustrated by the phylogeny tree with wild emmer as the outgroup. c, Evolutionary relationship of bread wheat and its wild progenitors in the genera Triticum and Aegilops. The sample size of the individual taxa was labeled. The chronogram of the phylogeny was obtained by calculating the divergence of orthologous genes between species (Methods and Supplementary Fig. 9). These high-coverage genomes (∼6.5×) empowered high-quality calling of genetic variations in self-pollinated plants such as wheats (Supplementary Table 3). By applying the cross-ploidy variation discovery pipeline (Supplementary Fig. 2)13, we identified ∼78 million single nucleotide polymorphisms (SNPs), and constructed version 1.1 of the whole-genome genetic variation map of wheat (VMap 1.1) (Supplementary Note 1, and Supplementary Fig. 3, and Supplementary Table 4 and 5). The false-positive error rate of variant calling, i.e., the proportion of segregating sites in the reference accession, Chinese Spring, was only 0.011%, which is similar to the error rates of high-quality SNPs in previous studies13,22. Spatiotemporal origin of bread wheat Many crops transited from weedy grasses to cultivated plants through solely domestication23. However, for bread wheat, this early transition was coupled with an additional polyploid speciation event, from which bread wheat arose through the hybridization between tetraploid wheats (AABB) and strangulata (Ae. tauschii ssp. strangulata, DD)24,25. Phylogenetic analyses of VMap 1.1 corroborated two recent findings regarding the origin of bread wheat (Fig. 1b,c, and Supplementary Note 2)13. One is the two-stage model of wheat domestication that wild emmer (T. turgidum ssp. dicoccoides, AABB) was transformed to domesticated emmer first, then free-threshing tetraploids. The other is the identification of free-threshing tetraploid wheats as the direct donor of the AB subgenomes during the polyploid speciation of bread wheat. Although the evolutionary topology of wheat populations becomes increasingly clear, there is limited consensus on the spatiotemporal dynamics of the emergence of bread wheat24. As the progenitor of domesticated emmer, wild emmer comprises two subpopulations mostly confined to the northern and southern Levant in West Asia (Fig. 2a)26,27. Archaeological records from early Neolithic sites showed that domesticated emmer appeared in the northern Levant (Abu Hureyra and Cafer Höyük) and southern Levant (Tell Aswad) almost simultaneously ∼9,800-9600 BP28, raising a controversial question in which place emmer wheat was first domesticated24. By reconstructing the phylogeny of AB lineage using 150,000 random SNPs, we found that wild emmer in the northern Levant was clustered with domesticated emmer (Fig. 1b). Moreover, bread wheat showed a closer identity by state (IBS) distance with northern wild emmer rather than southern wild emmer (Fig. 2a, and Supplementary Table 13 and 14). These results support the hypothesis that emmer wheat was domesticated around the Karacadag region in the northern Levant18,26. The birthplace of bread wheat is also mysterious. As the distribution of wild emmer and strangulata is primarily restricted to the Levant and the south of the Caspian Sea, respectively, it was puzzling how the polyploid speciation of bread wheat could occur given the geographic isolation of parental taxa24. Here we identified that free-threshing tetraploids rather than wild emmer were the donor of the AB subgenomes of bread wheat (Fig. 1b and c)13, suggesting the scenario that hexaploidization of bread wheat did not occur until free-threshing tetraploids expanded to the south of the Caspian Sea24. Further analyses of IBS distance showed that strangulata accessions in the southwest of the Caspian Sea have the greatest affinity to bread wheat (Fig. 2a, and Supplementary Table 15), indicating that bread wheat came into being at the southwest coast of the Caspian Sea25. |
|
|
|
Post by Admin on Mar 27, 2023 17:48:39 GMT
 Fig. 2 Demographic models of the bread wheat speciation. a, A geographic affiliation of IBS distances across bread wheat and its progenitors. Color scale indicates the distance of the AB subgenomes (blue) and the D subgenome (red) between bread wheat and progenitors. The map was created using the R package rworldmap. b, Timeline of evolutionary events related to bread wheat speciation. The top is the timeline of population split between wheats inferred from SMC++. The bottom is the wheat evolutionary timeline derived from archaeological evidence. c, The best supported demographic model of the speciation and introgression in wheats for AB subgenomes and D subgenome. The width of each grey rectangle indicates the estimated effective population size (Ne). Arrows among the grey rectangles are the migration rates (m) among different populations, and only 2Nem >1 is shown. The colored rectangle at the timeline indicates the time boundary of introgression. To provide a temporal context of wheat speciation, we used SMC++29, which combines the simplicity of sequentially Markovian coalescent and the scalability of site frequency spectrum (SFS) based approaches, to infer divergence time between wheat populations. Given the distinct evolutionary trajectories of the AB and D subgenomes of bread wheat (Fig. 1c), we inferred population split times of the AB and D lineages independently based on ∼68 million neutral SNPs in VMap 1.1 (Supplementary Note 3). The results from the AB lineage showed that domesticated emmer diverged from wild emmer 10,041±160 BP, free-threshing tetraploids separated from domesticated emmer 9,269±98 BP, and bread wheat split from free-threshing tetraploids 8,441±140 BP (Fig. 2b). The temporal sequence coincides nicely with the oldest archaeological remains of domesticated emmer6,28, free-threshing tetraploids30, and bread wheat24. Considering that hexaploidization of bread wheat involves free-threshing tetraploids and strangulata simultaneously (Fig. 1c), the speciation times of bread wheat inferred from the AB and D lineages should concur. However, we observed a drastic gap of ∼3,300 years between the two estimates, in which bread wheat diverged from strangulata 11,738±112 BP (Fig. 2b). Recent studies have identified an asymmetric wild-progenitor introgression in bread wheat, where introgression is much more prevalent in the AB subgenomes (19.43%) than in the D subgenome (0.49%)13. Given that gene flow can change the tempo of population differentiation31,32, the asymmetric introgression is likely to explain the different speciation times of bread wheat inferred from AB and D subgenomes. To provide a nuanced view of bread wheat speciation in the context of progenitor introgression, we investigated the chronology of gene flow between wheat populations through contrasting alternative demographic models33 (Fig. 2c, and Supplementary Note 4). By comparing the observed joint SFS of bread wheat and its progenitor population to the expected under a specific model, we found archaic gene flow from wild emmer and domesticated emmer into bread wheat before 8,919 BP (95% confidence interval (CI) 8,316-9,521 BP) and 7,228 BP (95% CI 6,760-7,695 BP), respectively. Moreover, the best-fitting model predicted enduring and bidirectional gene flow between free-threshing tetraploids and bread wheat since the emergence of bread wheat ∼11,700 BP. In contrast, the introgression from strangulata to the D subgenome of bread wheat was more ancient, predating 9,729 BP (95% CI 9,015-10,442 BP). These results suggest that the long-standing and massive gene flow to the AB subgenomes resulted in slow speciation of nascent bread wheat, lasting ∼3,300 years until the distinct genetic makeup of bread wheat was established. Notably, the near-complete reproductive isolation and concomitant clean-split between bread wheat and strangulata allow the estimate of the upper time-bound of population differentiation between cultivated crops and wild relatives, which is generally intractable in diploid crops. |
|
|
|
Post by Admin on Mar 28, 2023 19:23:05 GMT
Trans-Eurasian dispersal of bread wheat The spread of bread wheat across Eurasia profoundly transformed human societies10. To elucidate the range expansion process, we selected 225 bread wheat landraces (hereinafter referred to as landraces) from VMap 1.1 based on the accessibility of geographic information to characterize the spatiotemporal dispersal of bread wheat (Supplementary Table 18). Model-based clustering of landraces exhibited a salient east-west axis of range expansion of bread wheat originating from West Asia (Fig. 3a, and Supplementary Fig. 18), echoed by the Asian and European clades in the phylogeny of bread wheat (Fig. 1b). To reconstruct the bidirectional migration routes precisely, we applied the Estimated Effective Migration Surfaces (EEMS) method34 to identify spatial barriers and corridors of bread wheat expansion (Fig. 3a, and Supplementary Fig. 19). EEMS presented a fast migration route westward along the northern Mediterranean coast, consistent with the uniform ancestry of landraces in the area. In contrast, EEMS eastward migration patterns identified a massive roadblock at the Pamir Mountains that splits the Inner Asian landraces into Central and South Asian populations, suggesting the further spread of bread wheat eastward through the north and south routes of the Pamir Mountains.  Fig. 3 Trans-Eurasian expansion of bread wheat. a, Proposed dispersal routes of bread wheat in Eurasia. The map colors showed the estimated effective migration surfaces (EEMS) representing migration barriers (orange) and channels (cyan). Pies on the map showed the ancestral proportion of the five lineages. Arrows were the estimated migration routes from the Fertile Crescent to Europe and Asia. Boxes mark subpopulation hybridization and new subspecies formation events, and the stippled areas represent the regions where the hybridization events took place. b, Admixture graph model identifies the hybridization events of bread wheat in ten regions along the eastward route. Solid lines with arrowheads represent uniform ancestries, and attached numbers show scaled drift parameter f2. Dashed lines represent mixed ancestries, and attached values indicate estimated proportion of ancestry. c, Distribution of split times estimated from cross-coalescence analysis of different regions. The median and quartiles with whiskers reaching up to 1.5 times the interquartile range are shown in boxplots. d, Inheritance probability of four Triticum subspecies formed through hybridization during bread wheat dispersal. Landraces in East and South Asia exhibited a complex population structure, illustrating a convoluted population history of Asian bread wheat as suggested by recent archeological studies35–39. To disentangle the dispersal of bread wheat in the vast land of geographic and cultural diversity, especially how bread wheat spread into China, we used qpGraph40 to explore the relationships between local landrace populations defined by EEMS (Fig. 3b and Supplementary Table 19). By testing 61,214 candidate admixture graph models, the best-fitting graph (Z-score = -2.76) predicted three dispersal routes connecting Central and East Asia, coinciding with the postulated Southern Himalaya route38, Hexi Corridor route37–39, and Steppe route35,36, respectively. The Southern Himalaya route is from Pakistan, through India, Myanmar, and Yunnan Province, into China. The mixed ancestry of landraces in southwest China (R9) provided the first evidence demonstrating the existence of the southern route38. The Hexi Corridor route can also be referred to as “proto-silk Road,” starting from Central Asia, through the Inner Asian Mountain Corridor and Hexi Corridor to inner China. This route is the most prominent hypothesis describing wheat spread in China, verified by its abundant archaeological sites37–39. The Steppe route was recently proposed because the wheat remains excavated from the lower Yellow River region (∼4,250 BP) are earlier than those from the upper region (∼3,850 BP), indicating an alternative northern route to China via the Mongolian Steppe other than the Hexi Corridor35. Despite the lack of wheat samples from southern Mongolia, our results support this newly hypothesized route with genetic evidence—two populations in the lower Yellow River region (R4) and East China (R5) descended from past hybridization events (Fig. 3b), with one of the parental populations likely to be the lineage that traveled across the Mongolian Steppe. The introduction of wheat to China through the Mongolian Steppe may be related to early agropastoral societies, e.g., the Afanasievo people around the Altai Mountains, moving southward in response to the abrupt global cooling during the mid-Holocene36. We used SMC++29 to calculate splitting times between locally adapted and West Asia populations to infer the timing of bread wheat dispersal across Eurasia. Given that recent crop exchange and accompanying gene flow may reduce the divergence time estimates, we first assessed the temporal pattern of gene flow between individual local populations using fastsimcoal233. The results showed that populations in the Iberian Peninsula, Indus Valley, Yunnan Province, and East China exhibited early gene flow to the West Asia population (Supplementary Fig. 20 and Supplementary Table 20), and thus were qualified to calculate splitting times (Fig. 3c). As these four populations probably are not strictly locally confined, we inferred the timing of bread wheat dispersal at the continental level that bread wheat may have dispersed to Europe, South Asia, and East Asia ∼7,000 BP, ∼6,000 BP, and ∼5,400 BP, which are concordant with archeological records35,37,38. |
|
|
|
Post by Admin on Mar 29, 2023 18:36:02 GMT
New Triticum subspecies arising from bread wheat dispersal It becomes increasingly evident that interspecific hybridization is common during range expansion of species31. Bread wheat dispersal appeared to be no exception—we found several newly formed Triticum subspecies having their origins in sympatric hybridization between expanding bread wheat and locally preexisting tetraploid wheats. The phylogeny of Triticum populations showed that three hexaploid subspecies (AABBDD), including spelt (T. aestivum ssp. spelta), Macha (T. aestivum ssp. macha), and Xinjiang wheat (T. aestivum ssp. petropavlovskyi), were clustered into the tetraploid clade; similarly, a tetraploid subspecies (AABB), Persian wheat (T. turgidum ssp. carthlicum), was within the hexaploid clade (Fig. 1c and Supplementary Fig. 10). To clarify the ancestry of these outliers, we used phyloNet41 to infer reticulate phylogenetic networks of these subspecies based on phylogenies of 9,612 orthologous genes. The result showed a mixed ancestry of the four subspecies descending from hybrids between tetraploid wheats and bread wheat, with the genetic contribution of bread wheat from 33% to 54% (Fig. 3d and Supplementary Fig. 21). It is worth noting that spelt was considered the progenitor of bread wheat because it has a primitive phenotype of hulled seed42, our result indicates that the phenotype is inherited from its tetraploid parent, domesticated emmer, and thus disproves the once-popular theory concerning the origin of bread wheat. We then estimated the speciation time of the four subspecies using SMC++. To eliminate the noise from homoploid gene flow, we calculate the population splitting time between the hybrid offspring and only one of the parental taxa with different ploidy levels. The results showed spelt, Macha, Xinjiang wheat, and Persian wheat arose ∼6,400 BP, 7,300 BP, ∼3,300 BP, and ∼6,000 BP, respectively (Fig. 3a and Supplementary Fig. 22). By calculating the IBS distance between the four subspecies and individual accessions of their parental populations, we showed that these newly formed Triticum subspecies likely originate from Europe, West Asia, and Central Asia (Fig. 3a and Supplementary Fig. 23-26).  Fig. 4 Geographic expansion reshaped the adaptive genetic diversity of bread wheat. a, Landraces mapped on the first two canonical axes of Redundancy analysis (RDA). Arrows represent 20 environmental factors (11 temperature factors, 8 precipitation factors, and altitude) that are correlated with genotype of landraces. Colored points representing accessions from different regions: Europe (EU), West Asia (WA), Inner Asia (IA), East Asia (EA), and South Himalayas (SH). b, Ranked importance of environmental factors based on individual RDA analyses. c, Proportion of total SNP variance explained in RDA by environmental variable categories in each region. d, Sequence Ppd-D1 gene on the chromosome 2D of the reference genome (Chinese Spring). Three causative loss-of-function alleles and non-causative frameshift mutation are marked with red rectangles. The light-yellow rectangle represents the gene body. Blue rectangles represent exons. e, Selective sweeps on chromosome 2D to identify adaptive footprints on Ppd-D1. Top: IA vs. SH. Bottom: SH1 (Altitude > 3000 m) vs. SH2 (Altitude < 1000 m). The horizontal dotted lines indicate the top 5% genome-wide cut-off level. Arrows marked the position and top quantile of the Ppd-D1 gene. f, Haplotypes of Ppd-D1 gene in strangulata and bread wheat landrace. The numbers represent three loss-of-function genetic variants corresponding to d. The colored bars on the left represente different species/populations. g, Geographic distribution of the stop-gain mutation (number 2) of Ppd-D1 gene. h, Correlation between frequency and altitude of stop-gain mutation (number 2) of Ppd-D1 gene. i, Geographic distribution of ∼2kb deletion (number 1) of Ppd-D1 gene. j, Geographic distribution of 5-bp deletion (number 3) of Ppd-D1 gene. Orange indicates the proportion of three loss-of-function haplotypes in g, i and j, respectively. Geographic maps in g, i and j were created using the R package rworldmap. Genetic heritage of bread wheat expansion The trans-Eurasian dispersal of bread wheat may have involved extensive adaptive changes in the genome while colonizing novel environments. To investigate how the adaptation process affects the genetic diversity of bread wheat, we examined the correlations between SNPs and environmental variables of 225 landraces using redundancy analysis (RDA)43 (Supplementary Table 22). These environmental variables include altitude and 19 bioclimatic variables related to either temperature or precipitation. We found that these variables explained 13.44% of the total SNP variance. To evaluate the confounding effect of environmental adaptation and isolation-by-distance, we performed a similar RDA analysis using latitude and longitude, instead, as explanatory variables, finding that only 6.05% SNP variance was explained (Supplementary Fig. 27). The results demonstrate the importance of environmental factors in shaping the adaptive genetic diversity of bread wheat. By conducting individual RDA analyses on environmental variable categories, temperature-related variables (adjust r2 = 0.11) exhibited larger SNP variance than did precipitation (adjust r2 = 0.075) and altitude (adjust r2 = 0.013) (Fig. 4a). However, in search for the most important environmental variables, precipitation of the warmest quarter appeared on the top of the list (Fig. 4b), suggesting the complexity of local adaptation of bread wheat. To investigate the regional heterogeneity of adaptation, we performed RDA analyses on environmental variable categories using landraces from West Asia (WA), Europe (EU), Inner Asia (IA), East Asia (EA) and Southern Himalaya (SH) (Supplementary Fig. 28). The result showed that environment variables in WA explain the least SNP variance compared with other regions. In addition, the relative proportions of SNP variance explained by temperature, precipitation, and altitude varied in the five regions (Fig. 4c, Supplementary Fig. 29 and Supplementary Table 23). The results indicate that the accumulation of adaptive alleles from the range expansion has shaped a diverse adaptation landscape of bread wheat. To identify genomic regions associated with adaptation, we performed cross-population composite likelihood ratio (XP-CLR)44 analyses to detect selective sweeps between paired populations from the five populations mentioned above. Collectively, 185,865 selective sweeps were discovered under the top 5% XP-CLR score threshold. As these sweeps may stem from selections of human preference, farming practices, etc., we then conducted environmental association analyses using Bayenv45 to narrow down the candidate sweep regions to those related to environmental factors. Based on associations between 20 environmental variables and allele frequency of 1.5M SNPs in 13 populations (Supplementary Fig. 30-32 and Supplementary Table 24), the analysis identified 269,279 adaptation-associated SNPs (top 5% Bayes factor) intersecting with selective sweeps from XP-CLR, with an average of 2.15-fold enrichment for coexisting with sweep regions (Supplementary Fig. 33 and Supplementary Table 25). A total of 19,999 genes were identified as being involved in the environmental adaptation of bread wheat, including 123 cloned genes that regulate critical agronomic traits, such as disease resistance and abiotic stress response, etc. (Supplementary Fig. 34, and Supplementary Table 26-28), indicating the value of adaptation-associated genes in improving agronomic traits of modern wheat. |
|

