|
|
Post by Admin on Dec 31, 2020 19:43:15 GMT
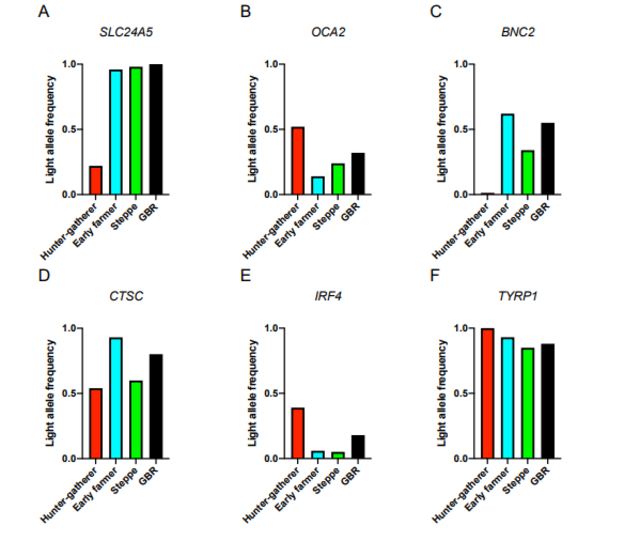 Fig. S6. Light allele frequencies in ancient and present-day European populations for select SNPs (A) rs2675345, (B) rs4778123, (C) rs2153271, (D) rs3758833, (E) rs12203592, and (F) rs1325132. Nearest genes are labelled at the top of each bar plot. In the supplementary section of this paper (Ju and Mathieson 2021), OCA2 and TYRP1 are identified as light skin alleles for WHGs. Their frequencies are 50% and 100%, respectively. There are several other pigmentation genes attributed to WHGs such as CTSC (55%) and IRF4 (40%). The IRF4 gene is strongly associated with pigmentation and sensitivity of skin to sun exposure. Previous papers on human pigmentation (Günther et al. 2018) had not found these pigmentation genes in WHGs. Figure 4 shows that 100% of WHGs had the OCA2/HERC2 gene, which is associated with blue eye color and light skin (Günther et al. 2018). However, most studies only link this OCA2/HERC2 gene to blue eyes, concluding that WHGs had blue eyes and dark skin. The OCA2 gene is independently involved in the evolution of light skin pigmentation in East Asia and Europe. Two OCA2 polymorphisms (rs1800414 and rs74653330) have been associated with lighter skin pigmentation in East Asian populations. Aside from three genes associated with light skin in Europe (SLC24A5, SLC45A2 and TYRP1) which began to increase in frequency between 19,000 and 11,000 years ago, OCA2 also showed a potential signal of selection in Europeans and played part in light skin pigmentation in Europe. WHGs had fewer depigmentation genes compared to modern Europeans but they were probably as light-skinned as East Asians. 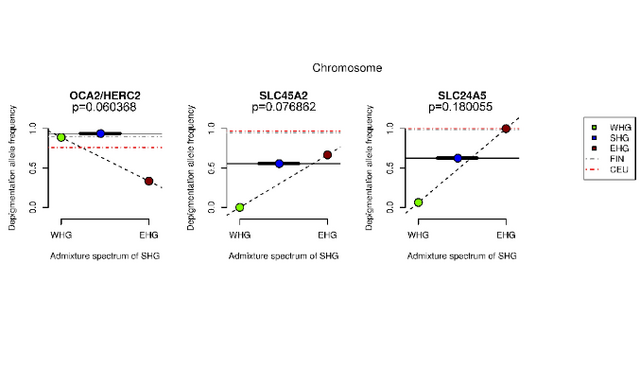 Fig 4. Adaptation to high-latitude environments. Discussion Large whole-genome ancient DNA datasets have, in the past few years, allowed us to track the evolution of variation associated with both simple and complex traits (68, 69). The majority of samples are from Western Eurasia, and we focus on that region, noting that a parallel process of selection for light skin pigmentation has acted in East Asian populations (9, 70). In West Eurasia, selection for skin pigmentation appears to have been dominated by a small number of selective sweeps at large-effect variants. There are a number of possible explanations for this. Many of the variants detected by the UK Biobank GWAS may have such small effects that they were effectively neutral or experienced such weak selection that we cannot detect it. Other variants may not have responded to selection because of pleiotropic constraint. The five SNPs with the largest effect sizes in UK Biobank do have some have small but significant associations with anthropometric phenotypes (for example, rs1805007 at MC1R with standing height), but by far their most significant associations are with hair color, facial aging, tanning, melanoma risk, and other phenotypes that are likely related to pigmentation (64). Finally, GWAS effect size estimates in European populations today may not reflect the impact these variants had in the past due to epistatic or gene–environment interactions. This would not affect the individual SNP time series and PBS analyses since they do not rely on effect sizes as weights but would reduce power for the weighted polygenic score and Qx analyses. Our analyses centered on 170 skin pigmentation-associated SNPs that are present on the 1240K capture array. To check for possible bias introduced by the capture array, we checked how many of the 242 SNPs used in the shotgun analysis were well tagged by the capture array. Of the 242 SNPs, 33 are shared, a further 67 are tagged at r2 ≥ 0.8, and a further 19 SNPs are tagged at r2 ≥ 0.5. However, while the 142 SNPs that are not tagged at r2 ≥ 0.8 by the capture array do contain some moderate effect size SNPs, they do not contribute to the selection signal (P = 0.31) (SI Appendix, Fig. S15D). Therefore, although the capture array does not capture all of the variation contributing to present-day skin pigmentation variation, it does capture the vast majority of the variation that contributed to the evolution of the phenotype, justifying the use of the capture-shotgun dataset for detailed investigation of the evolutionary trends. We find little evidence of parallel selection on independent haplotypes at skin pigmentation loci, suggesting that that differences in allele frequency across ancestry groups were mostly due to genetic drift. One exception is that the light allele at SLC24A5 was nearly fixed in both Early Farmer and Steppe ancestry populations due to selection. However, even for this variant we observe a signal of ongoing selection in our data even after admixture with hunter-gatherers, indicating continued selection after admixture. This is analogous to the rapid selection at the same locus for the light allele introduced via admixture into the KhoeSan, who now occupy southern Africa (12, 71). We are also able to test previous claims about selection on particular pigmentation genes. We find no evidence of positive selection in Europeans at the MC1R locus in contrast to previous reports (72, 73). Among UK Biobank SNPs, rs1805007 near MC1R explains a relatively large amount of variation within the UK but is predicted to explain relatively little of the variation between Europe and West Africa (Fig. 4C). The TYRP1 locus has been previously identified as a target of selection in Europeans (5, 8, 9, 74, 75), although some studies (76, 77) have questioned this finding. Our analysis shows some support for selection at this locus, with a 20-SNP window centered around rs1325132 being in the top 1.1% of genome-wide PBS windows on the hunter-gatherer lineage. Some studies (76, 78) detect a signal of recent selection in Europeans at the KITLG SNP rs12821256 that is functionally associated with blond hair color (79). We find no evidence of selection on the West Eurasian lineage at this SNP in our time series analysis, but this may reflect a lack of power to detect a relatively small change in frequency. Finally, at the OCA2 locus, we recapitulate an observation of independent selection in Europeans and East Asians (70, 80) (SI Appendix, Fig. S9). In the ancient populations, we found an elevated PBS signal around rs9920172 that was most prominent in hunter-gatherers but also elevated in Early Farmer and Steppe ancestry populations (Fig. 3), suggesting a relatively early episode of selection in West Eurasians. Relatively dark skin pigmentation in Early Upper Paleolithic Europe would be consistent with those populations being relatively poorly adapted to high-latitude conditions as a result of having recently migrated from lower latitudes. On the other hand, although we have shown that these populations carried few of the light pigmentation alleles that are segregating in present-day Europe, they may have carried different alleles that we cannot now detect. As an extreme example, Neanderthals and the Altai Denisovan individual show genetic scores that are in a similar range to Early Upper Paleolithic individuals (SI Appendix, Table S1), but it is highly plausible that these populations, who lived at high latitudes for hundreds of thousands of years, would have adapted independently to low UV levels. For this reason, we cannot confidently make statements about the skin pigmentation of ancient populations. Our study focused, for reasons of data availability, on the history of skin pigmentation evolution in West Eurasia. However, there is strong evidence that a parallel trend of adaptation to low UVB conditions occurred in East Asia (15, 70, 81, 82). Less is known about the loci that have been under selection in East Asia, aside from some variants at OCA2 (80⇓–82). Similarly, multiple studies have documented selection for both lighter and darker skin pigmentation in parts of Africa (12, 13, 83). Future work should test whether the process of adaptation in other parts of the world was similar to that in Europe. The lack of known skin pigmentation loci in scans of positive selection in East Asian populations (84, 85) raises the possibility that selection may have been more polygenic in East Asians than in Europeans. Finally, the evidence for polygenic directional selection on other complex traits in humans is inconclusive (86, 87). We suggest that detailed studies of other phenotypes using ancient DNA can be helpful at more generally identifying the types of processes that are important in human evolution. |
|
|
|
Post by Admin on Jan 6, 2021 20:40:13 GMT
 About a year and a half ago at ASHG, I had a discussion with Dan Ju and Iain Mathieson about their work on ancient pigmentation. Or, more precisely, ancient pigmentation related genes. Now it’s out in a preprint, The evolution of skin pigmentation associated variation in West Eurasia: …It is unclear whether selection has operated on all the genetic variation associated with skin pigmentation as opposed to just a small number of large-effect variants. Here, we address this question using ancient DNA from 1158 individuals from West Eurasia covering a period of 40,000 years combined with genome-wide association summary statistics from the UK Biobank. We find a robust signal of directional selection in ancient West Eurasians on skin pigmentation variants ascertained in the UK Biobank, but find this signal is driven mostly by a limited number of large-effect variants. Consistent with this observation, we find that a polygenic selection test in present-day populations fails to detect selection with the full set of variants; rather, only the top five show strong evidence of selection. Our data allow us to disentangle the effects of admixture and selection. Most notably, a large-effect variant at SLC24A5 was introduced to Europe by migrations of Neolithic farming populations but continued to be under selection post-admixture. This study shows that the response to selection for light skin pigmentation in West Eurasia was driven by a relatively small proportion of the variants that are associated with present-day phenotypic variation.  There are a lot of moving parts in this preprint. Look closely, and you will notice that the authors are careful to stipulate that they can’t really infer the pigmentation of ancient peoples, only the alleles ascertained in modern populations. This matters, because naive deployments of polygenic risk score models trained on modern populations projected on ancient ones seem highly suspect. I’m thinking here mostly of the “Cheddar Man is black” meme. It is true that using modern SNP batteries Mesolithic Europeans are predicted to be rather dark-skinned, but higher latitude humans tend to be paler, on average, than lower latitude humans (albeit, not as pale as the typical Northern European!). But, we can be sure about the alleles we do know about, and, their likely effect (the functional understanding of these pathways is pretty good). The best modern genetic analyses of pigmentation suggest that variation is dominated by some large-effect loci, but that there is a large residual of smaller-effect loci segregating within the population (I’ve seen 50% accounted for with SNPs, and 50% as “ancestry”, which really masks small-effect QTLs). This is in contrast with the architecture in height, where there are few large-effect loci, and almost all of the variance is small-effect loci. What Ju et al. confirm is that selection “for pigmentation” is due to the large-effect loci; there’s no polygenic selection detectable on the smaller-effect loci for the ancient populations. Importantly, the change in allele frequency isn’t just due to admixture. It’s also due to selection after admixture. I use quotes above because honestly, I think these sorts of results make it unclear what the selection was for. The general prior is conditioned on the fact that even after a few decades we still think of EDAR as a hair-thickness gene, but it’s one of the strongest signals of selection in the human genome. The “light” allele in SLC24A5 is at an incredibly high frequency in Europe today, and has increased in the last 4,000 years. Though this SNP is impactful for the complexion, it’s hard to imagine how strong selection must be to drive it from 95% to 99.5% (as per 2005 paper on this SNP, the “light” allele exhibits some phenotypic dominance). As noted in the preprint, there’s not enough data on other regions of the world. It’s hard to assess what’s going on Europe without assessing other regions. The authors do present an intriguing suggestion: that lighter pigmentation in East Asia is driven by smaller-effect genes shifted through polygenic selection. I’ll present a strange hypothesis: selection for lighter skin at high latitudes through polygenic selection on standing variation naturally takes populations to the coloring of Northeast Asians. But very light complexion, as you see in Northern Europe, could be due to strong selection on the large-effect pigmentation genes, and pigmentation itself may simply be a side effect due to a genetic correlation with the true target of selection. |
|
|
|
Post by Admin on Jan 7, 2021 21:01:30 GMT
Understanding the genetic basis of pigmentation in human populations Pigmentation shows a tremendous amount of variation in human populations, most probably due to the action of natural selection. In spite of decades of study, there are many gaps in our knowledge of the genetic basis of normal pigmentation variation and the evolutionary history of the genes responsible for pigmentation. One of the main reasons for this lack of knowledge is the multifactorial nature of this trait. Although the last century has witnessed impressive advances in our understanding of simple Mendelian traits and diseases, elucidating the genetic factors responsible for variation in complex traits remains a challenge. However, conditions are in place to identify the ultimate genetic factors involved in pigmentation variation within and between populations. We have better technologies and a better knowledge of the pigmentary system than anytime in the past, and there is much that can be learned from a systematic study of pigmentation candidate genes in multiple population groups. To date, most of the studies focusing on the genetics of pigmentation have been carried out in populations of European ancestry, so it is critical to expand this research to other population groups.  In 2010, we described that a non-synonymous polymorphism in the OCA2 gene (rs1800414) is significantly associated with skin pigmentation in East Asians (Edwards et al. 2010. PLoS Genet. 6:e1000867). This was one of the first polymorphisms associated with skin pigmentation in East Asian populations. More recently, we described that another non-synonymous variant in the OCA2 gene (rs74653330) also shows a strong association with skin pigmentation in East Asians (Eaton et al. 2015. Am. J. Hum Biol. 27:520-525). Interestingly, our research shows that the OCA2 gene has been independently involved in the evolution of pigmentation in Europe and East Asia. In Europe, haplotypes in the OCA2/HERC2 gene are associated with blue eye color and light skin, and these haplotypes are not present in East Asian populations. On the other side, the rs1800414 G and the rs74653330 A alleles, which are associated with light skin in East Asia, are very rare or absent in other population groups. This is one of the most fascinating examples of convergent evolution in our species. We have also tackled another fascinating research question: When did the reduction in melanin levels take place in Europe and East Asia? In order to do this, we estimated the age of some of the key alleles associated with pigmentation in both continents. In collaboration with Sandra Beleza (Stanford University) and Jorge Rocha (Universidade do Porto) we estimated that the alleles of three genes associated with light skin in Europe (SLC24A5, SLC45A2 and TYRP1) began to increase in frequency between 19,000 and 11,000 years ago, possibly coinciding with population size expansions in Europe (Beleza et al. 2012. Mol. Biol. Evol. 30:24-35). We used a different method to date the two OCA2 alleles associated with pigmentation in East Asia. Our estimates of the Most Recent Common Ancestor (MRCA) were under 10,000 years ago for both alleles (Murray et al. 2015. Hum Genome Var. 2:15058). Therefore, it seems that the major events involved in the lightening of skin pigmentation in Europe and East Asia took place long after the arrival of anatomically modern humans to these regions (around 45,000 years ago). Recent ancient DNA studies in Europe tend to support these findings. Similar studies in East Asia will provide relevant information about the timing of the selective events in this continent.  We are also interested in the development of quantitative methods to study pigmentary phenotypes, including iris color. This is important, because the categorical classifications widely used to classify iris color (e.g. brown, blue, green) do not capture the continuous nature of this trait. In a paper published in 2012 we described an automated method to quantify iris color using high-resolution pictures. (Edwards et al. 2012. Am. J. Phys. Anthropol. 147:141-149). More recently, we improved the method to provide, not only information about iris color, but also about different surface features present in the iris, such as Fuchs crypts, Wolfflin nodules, pigment spots, contraction furrows and conjunctival melanosis (Edwards et al. 2016. Pigment Cell Melanoma Res. in press, and Edwards et al. 2016. R. Soc. Open Sci. 3:150470). Through the years, we have done many candidate gene association studies focused on pigmentation in multiple population groups (see list of references for additional information). Currently, in collaboration with Heather Norton (University of Cincinnati), we are involved in a genome-wide association study of pigmentary traits and iris surface features in five population groups: African Americans, East Asians, Europeans, Hispanics and South Asians. We hope that this study, using quantitative approaches to describe the relevant traits, will provide interesting insights about the genetic architecture of pigmentary traits in these populations. |
|
|
|
Post by Admin on Jan 17, 2021 20:54:38 GMT
The distinctive geographic patterns of common pigmentation variants at the OCA2 gene
Kenneth K. Kidd, Andrew J. Pakstis, Michael P. Donnelly, Ozlem Bulbul, Lotfi Cherni, Cemal Gurkan, Longli Kang, Hui Li, Libing Yun, Peristera Paschou, Kelly A. Meiklejohn, Eva Haigh & William C. Speed
Scientific Reports volume 10, Article number: 15433 (2020)
Abstract
Oculocutaneous Albinism type 2 (OCA2) is a gene of great interest because of genetic variation affecting normal pigmentation variation in humans. The diverse geographic patterns for variant frequencies at OCA2 have been evident but have not been systematically investigated, especially outside of Europe. Here we examine population genetic variation in and near the OCA2 gene from a worldwide perspective. The very different patterns of genetic variation found across world regions suggest strong selection effects may have been at work over time. For example, analyses involving the variants that affect pigmentation of the iris argue that the derived allele of the rs1800407 single nucleotide polymorphism, which produces a hypomorphic protein, may have contributed to the previously demonstrated positive selection in Europe for the enhancer variant responsible for light eye color. More study is needed on the relationships of the genetic variation at OCA2 to variation in pigmentation in areas beyond Europe.
Introduction
Oculocutaneous Albinism type 2 (OCA2) is a gene of interest for several reasons, not the least of which is its role in oculocutaneous albinism with about 30% of worldwide cases accounted for by 154 mutations in the OCA2 gene1. Two amino acid substitutions in the coding sequence were shown by Sviderskaya et al.2 to be associated with decreased expression of the OCA2 protein but not full ocular albinism. OCA2 was subsequently studied for its association with eye color but common variants are associated not just with variation in eye color but also with variation in skin color3,4,5. Different polymorphisms in the regulatory and coding regions are primarily associated with different eye, hair, and skin pigmentation phenotypes and show large frequency differences among populations from different parts of the world.
Single nucleotide polymorphisms (SNPs) in the molecular region of OCA2 were first implicated in inheritance of eye color variation in Europeans6. The strongest evidence was for variation upstream of the OCA2 coding sequences in one of the introns of HERC27, supported by broader population genetics studies8. Sturm et al.7 showed that rs12913832 disrupted a conserved regulatory region; the region was subsequently confirmed to be an enhancer of OCA29. Functional variation in the HERC2 coding sequences seems unrelated to eye color7. OCA2 also has four commonly occurring SNPs that cause amino acid substitutions: rs1800414 (His615Arg), rs74653330 (Ala481Thr), rs1800407 (Arg419Gln), and rs1800401 (Arg305Trp). The Ala481Thr (rs74653330) and Val443Ile (rs121918166) variants were shown2 to be hypomorphic but not pathogenic in their studies of ocular albinism. The Val443Ile missense variant (rs121918166) has been reported at < 1% in Scandinavian populations10. These missense SNPs are distributed across 63 kb of the gene (Table 1); the enhancer SNP (rs12913832) is 38.6 kb from the start of the coding sequence.
Table 1 Five commonly occurring and one rare functional SNP at OCA2 influencing expression of human pigmentation variation.
Nucleotide position GRCh38 Distance to next SNP in basepairs dbSNP rs-number Ancestral amino acids‡ Anc-position#-Drv Alleles Anc, Drv forward strand Hypomorphic
27,951,891 31,516 rs1800414 His615Arg T,C
27,983,407 1,694 rs74653330 Ala481Thr C,T mildly
27,985,101 71 rs121918166 * Val443Ile C,T very
27,985,172 29,735 rs1800407 Arg419Gln C,T possibly
28,014,907 66,967 rs1800401 * Arg305Trp G,A
28,081,874 38,598 – initiation codon –
28,120,472 rs12913832 enhancer A,G
*SNPs not studied in this report.
‡Anc, ancestral; Drv, derived; amino acids–Ala, alanine; Arg, arginine; Gln, glutamine; His, histidine; Ile, isoleucine; Thr, threonine; Trp, tryptophan; Val, valine.
|
|
|
|
Post by Admin on Jan 18, 2021 4:14:31 GMT
Three of the OCA2 missense SNPs (rs1800414, rs74653330, rs1800407) have been studied in conjunction with pigmentation phenotypes, primarily in European and East Asian populations where the variants are most common. Walsh et al.11,12,13 found that including the genotype at rs1800407 in a regression equation improved the ability to predict eye color in their samples. Edwards et al.14 and Yuasa et al.15,16,17 found that rs1800414 was associated with skin color variation among individuals of East Asian ancestry. Eaton et al.18 studied both rs1800414 and rs74653330 on East Asians and found them to be independently associated with skin color. Rawofi et al.19 confirmed the association of rs1800414 with skin color and found it significantly associated with iris color. Lee et al.20 identified the derived allele at rs74653330 at a frequency of about 1% in Europeans. This hypomorphic OCA2*481Thr (rs74653330) allele was later found to be moderately frequent in many East Asian populations17,21.
Evidence of recent selection for the derived allele of rs12913832 at the enhancer is clearly documented in European populations as is selection for the derived allele at rs1800414 in East Asia8. The skin color effects of rs1800414 have been considered an example of parallel evolution for light skin color14. We are interested in these and other aspects of the population genetics of the OCA2 variants. To that end we have tested (Table 1) four of the functional SNPs in the large number of population samples we have available22. We have also retrieved data on these SNPs from the 1,000 Genomes (1 KG) project website23 in those populations and assembled the published data on population frequencies. The derived alleles show very distinct biogeographic variation. That global pattern of variation is the focus of this paper.
Methods
Markers and Populations
Table 1 lists the three amino acid substitution SNPs at OCA2, rs1800414, rs74653330, and rs1800407 and the OCA2 enhancer SNP, rs12913832, in an intron of HERC2, that are the focus of this study. Data on all four of these SNPs come primarily from our genotyping studies (76 populations), from a collaboration with co-author Longli Kang (7 populations), and from the 22 relatively unadmixed populations of the 1 KG project (Phase 3)23. Additional individual SNP frequencies were obtained primarily from the published literature and were entered into the ALFRED database (https://alfred.med.yale.edu) before it became static. A fourth amino acid substitution, rs1800401 (Arg305Trp), has been typed in the 1 KG samples but is not included here because it has been otherwise studied largely in samples defined by pigmentation phenotypes (eye, hair, skin color) in a few populations3,24,25. The rare amino acid change (Val443Ile) at SNP rs121918166 has only been studied on a small number of European populations10 and studied for its effect on eye, hair, and skin color. Only three of the 1 KG populations, all European, have the variant allele at rare frequencies ranging from 0.5% to 0.9%. All of the samples were collected with informed consent for population genetic studies such as this. Because all samples are completely anonymous, the allele frequency collection in this study is not considered human research.
Marker Typing
Various methods were used to type the SNPs and are described in the multiple sources of the data. The source of data for each population sample is listed in Table S1 of supplemental data. The populations typed in Kidd Lab as part of this study were typed using TaqMan SNP Genotyping Assays obtained from Applied Biosystems as previously described; data on some of the SNPs in some of the populations were previously published8,26.
Statistics
As these SNPs are simple co-dominant genetic systems allele frequencies were estimated by simple gene counting. The density plots were produced by Surfer (version 12.8) software (https://www.goldensoftware.com). The haplotype frequencies were estimated using Phase version 2.1.127,28. Each population was phased separately.
|
|

