|
|
Post by Admin on Feb 14, 2020 19:01:49 GMT
 The study indicated that present-day West Africans trace a substantial proportion, some 2% to 19%, of their genetic ancestry to an extinct human species – what the researchers called a "ghost population." "We estimate interbreeding occurred approximately 43,000 years ago, with large intervals of uncertainty," said University of California, Los Angeles (UCLA) human genetics and computer science professor Sriram Sankararaman, who led the study published this week in the journal Science Advances. Homo sapiens first appeared a bit more than 300,000 years ago in Africa and later spread worldwide, encountering other human species in Eurasia that have since gone extinct including the Neanderthals and the lesser-known Denisovans. Previous genetic research showed that our species interbred with both the Neanderthals and Denisovans, with modern human populations outside of Africa still carrying DNA from both. But while there is an ample fossil record of the Neanderthals and a few fossils of Denisovans, the newly identified "ghost population" is more enigmatic. 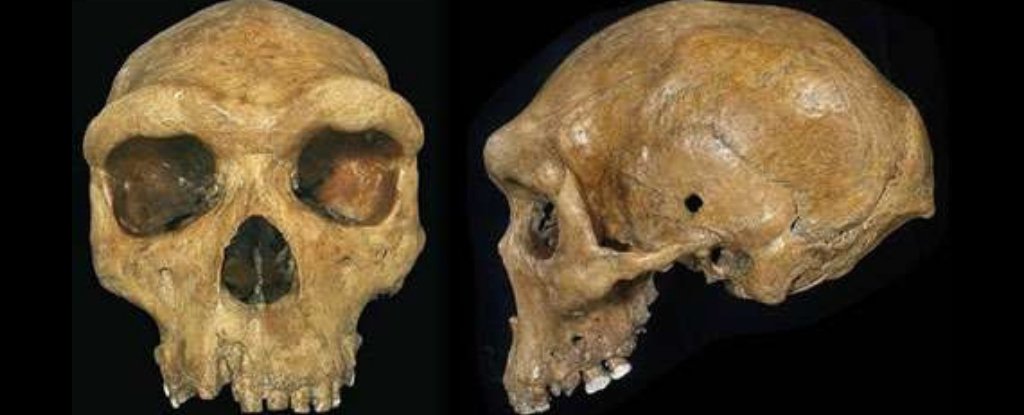 Asked what details are known about this population, Sankararaman said, "Not much at this stage." "We don't know where this population might have lived, whether it corresponds to known fossils, and what its ultimate fate was," Sankararaman added. Sankararaman said this extinct species seems to have diverged roughly 650,000 years ago from the evolutionary line that led to Homo sapiens, before the evolutionary split between the lineages that led to our species and to the Neanderthals. The researchers examined genomic data from hundreds of West Africans including the Yoruba people of Nigeria and Benin and the Mende people of Sierra Leone, and then compared that with Neanderthal and Denisovan genomes. They found DNA segments in the West Africans that could best be explained by ancestral interbreeding with an unknown member of the human family tree that led to what is called genetic "introgression." It is unclear if West Africans derived any genetic benefits from this long-ago gene flow. "We are beginning to learn more about the impact of DNA from archaic hominins on human biology," Sankararaman said, using a term referring to extinct human species. "We now know that both Neanderthal and Denisovan DNA was deleterious in general but there were some genes where this DNA had an adaptive impact. For example, altitude adaptation in Tibetans was likely facilitated by a Denisovan introgressed gene." |
|
|
|
Post by Admin on Feb 15, 2020 19:51:52 GMT
Abstract While introgression from Neanderthals and Denisovans has been documented in modern humans outside Africa, the contribution of archaic hominins to the genetic variation of present-day Africans remains poorly understood. We provide complementary lines of evidence for archaic introgression into four West African populations. Our analyses of site frequency spectra indicate that these populations derive 2 to 19% of their genetic ancestry from an archaic population that diverged before the split of Neanderthals and modern humans. Using a method that can identify segments of archaic ancestry without the need for reference archaic genomes, we built genome-wide maps of archaic ancestry in the Yoruba and the Mende populations. Analyses of these maps reveal segments of archaic ancestry at high frequency in these populations that represent potential targets of adaptive introgression. Our results reveal the substantial contribution of archaic ancestry in shaping the gene pool of present-day West African populations. INTRODUCTION Admixture has been a dominant force in shaping patterns of genetic variation in human populations (1). Comparisons of genome sequences from archaic hominins to those from present-day humans have documented multiple interbreeding events, including gene flow from Neanderthals into the ancestors of all non-Africans (2), from Denisovans into Oceanians (3) and eastern non-Africans (4, 5), as well as from early modern humans into the Neanderthals (6). However, the sparse fossil record and the difficulty in obtaining ancient DNA have made it challenging to dissect the contribution of archaic hominins to genetic diversity within Africa. While several studies have revealed contributions from deep lineages to the ancestry of present-day Africans (7–12), the nature of these contributions remains poorly understood. RESULTS We leveraged whole-genome sequence data from present-day West African populations and archaic hominins to compute statistics that are sensitive to introgression in the history of these populations. Specifically, we tabulated the distribution of the frequencies of derived alleles (where a derived allele is determined relative to an inferred human ancestor) in the analyzed African populations at single-nucleotide polymorphisms (SNPs) for which a randomly sampled allele from an archaic individual was observed to also be derived. Theory predicts that this conditional site frequency spectrum (CSFS) is expected to be uniformly distributed when alleles are neutrally evolving under a demographic model in which the ancestor of modern and archaic humans, assumed to be at mutation-drift equilibrium, split with no subsequent gene flow between the two groups (13, 14). This expectation is robust to assumptions about changes in population sizes in the history of modern human or archaic populations. Further, we show that this expectation holds even when there is population structure or gene flow in the history of the archaic population (see Materials and Methods). We computed CSFSYRI,N: the CSFS in the Yoruba from Ibadan (YRI) while restricting to SNPs where a randomly sampled allele from the high-coverage Vindija Neanderthal (N) genome was observed to be derived (15). In contrast to the uniform spectrum expected from theory, we observe that the CSFSYRI,N has a U-shape with an elevated proportion of SNPs with low- and high-frequency–derived alleles relative to those at intermediate frequencies (Fig. 1 and fig. S4). The CSFS is nearly identical when we replace the Vindija Neanderthal genome with the high-coverage Denisova genome (Fig. 1 and fig. S4) (4). We observed a similar U-shaped CSFS in each of three additional West African populations [Esan in Nigeria (ESN), Gambian in Western Divisions in the Gambia (GWD), and Mende in Sierra Leone (MSL)] included in the 1000 Genomes Phase 3 dataset (fig. S4).  Fig. 1 Demography relating known and proposed archaic lineages to modern human populations. (A) Basic demographic model with CSFS fit. W Afr, West Africans; Eur, European; N, Neanderthal; D, Denisovan; UA, unknown archaic [see (18)]. Below, we show the CSFS in the West African YRI when restricting to SNPs where a randomly sampled allele from the high-coverage Vindija Neanderthal was observed to be derived [Neanderthal (data)], as well as where a randomly sampled allele from the high-coverage Denisovan genome was observed to be derived [Denisovan (data)]. We also show the CSFS under the proposed model [Neanderthal (model) and Denisova (model)]. Migration between Europe and West Africa introduces an excess of low-frequency variants but does not capture the decrease in intermediate frequency variants and increase in high-frequency variants. (B) Newly proposed model involving introgression into the modern human ancestor from an unknown hominin that separated from the human ancestor before the split of modern humans and the ancestors of Neanderthals and Denisovans. Below, we show the CSFS fit from the proposed model, which captures the U-shape observed in the data. Mutational biases, errors in determining either the ancestral or the archaic allele, or recurrent mutation could produce the observed CSFS. We confirmed that the shape of the CSFSYRI,N was robust to the inclusion of only transition mutations, only transversion mutations, to the exclusion of hypermutable CpG sites (fig. S7), as well as when we computed the spectrum on the Yoruba genomes separately sequenced in the 1000 Genomes Phase 1 dataset (fig. S7). We verified that this signal was robust to changes in recombination rate and background selection by restricting to regions that are likely to be evolving neutrally (by restricting to sites with estimates of background selection, B statistic, >800). We also assessed the effect of biased gene conversion by excluding weak-to-strong and strong-to-weak polymorphisms. We found that the U-shaped signal is robust to variation in recombination rate, background selection, and biased gene conversion (fig. S10). Errors in determining the ancestral allele could make low-frequency ancestral alleles appear to be high-frequency–derived alleles and vice versa and thus could potentially lead to a U-shaped CSFS. However, the shape of the CSFS remains qualitatively unchanged when we used either the chimpanzee genome or the consensus across the orangutan and chimpanzee genomes to determine the ancestral allele (fig. S9). We simulated both ancestral allele misidentification and errors in genotype calling in the high-coverage archaic genome. A fit to the data required both a 15% ancestral misidentification rate and a 3% genotyping error rate in the archaic genome, substantially larger than previous estimates of these error rates [1% for ancestral misidentification rate in the Enredo-Pecan-Ortheus (EPO) ancestral sequence (16) and 0.6% for the modern human contamination in the Vindija Neanderthal (15)] (section S1.1 and fig. S11). To explore the contribution of recurrent mutations, we used forward-in-time simulations that allow for recurrent mutations: The simulated CSFS does not resemble the U-shaped CSFS that we see in data (fig. S43). Together, these results indicate that the U-shaped CSFS observed in the African populations is not an artifact. |
|
|
|
Post by Admin on Feb 16, 2020 6:18:32 GMT
To determine whether realistic models of human history can explain the CSFS, we compared the CSFS estimated from coalescent simulations to the observed CSFSYRI,N using a goodness-of-fit test (see Materials and Methods and section S2). We augmented a model of the demographic history of present-day Africans (17) with a model of the history of Neanderthals and Denisovans inferred by Prüfer et al. (15) (Fig. 1 and figs. S1 and S16). This model includes key interbreeding events between Neanderthals, Denisovans, and modern human populations such as the introgression from Neanderthals into non-Africans, from early modern humans into Neanderthals (6), and into the Denisovans from an unknown archaic population (18). The resulting model fails to fit the observed CSFSYRI,N . Extensions of this model to include realistic variation in mutation and recombination rates along the genome (KS P < 2 × 10−16; fig. S12 and section S1) and low levels of Neanderthal DNA introduced into African populations via migration between Europeans and Africans do not provide an adequate fit (KS P < 2 × 10−16; Fig. 1 and section S1) nor does a model of gene flow between YRI and pygmy populations that has been proposed previously (KS P < 2 × 10−16; fig. S12 and section S1) (19). The expectation that the CSFS is uniformly distributed across allele frequencies relies on an assumption of mutation-drift equilibrium in the population ancestral to modern humans, Neanderthals, and Denisovans. We confirmed that violations of this assumption (due to bottlenecks, expansions, and population structure in the ancestral population) were also unable to fit the data (KS P < 2 × 10−16 for all models; section S2, table S3, and fig. S17).
Given that none of the current demographic models are able to fit the observed CSFS, we explored models where present-day West Africans trace part of their ancestry to (A) a population that split from their ancestors after the split between Neanderthals and modern humans, (B) a population that split from the ancestor of Neanderthals after the split between Neanderthals and modern humans, or (C) a population that diverged from the ancestors of modern humans and Neanderthals before the ancestors of Neanderthals and modern humans split from each other (fig. S2 and section S3). Each of these models of admixture (which we refer to as models A, B, and C, respectively) can yield a U-shaped CSFS. The increase in the counts of low derived allele frequency SNPs is largely due to the introduction of the derived allele from the introgressing population at sites that are fixed for the ancestral allele. The increase in the counts of the high-frequency SNPs is largely due to the introduction of the ancestral alleles at sites that are fixed for the derived allele.
A search for the parameters for models A and B that produce the best fit to the CSFS results in a trifurcation, i.e., models in which the introgressing population splits off from the modern human population at the same time as the modern human–Neanderthal. Models A and B fail to fit the observed CSFS even at their most likely parameter estimates (KS P = 3.3 × 10−15 and P = 5.6 × 10−6, respectively; section S3) because of insufficient genetic drift in the African population since the split from the introgressing population (section S4.2). In addition, we show in appendix B that the spectrum for model A is expected to be symmetric, which is not observed in the data (Fig. 1). Model C, on the other hand, is consistent with the data (KS P = 0.09), suggesting that part of the ancestry of present-day West Africans must derive from a population that diverged before the split time of Neanderthals and modern humans. In addition to the goodness-of-fit tests, we examined the likelihood of the best-fit parameters for each of the models and found that model C provides a significantly better fit than other models (model C having a higher composite log likelihood than the next best model Δℒℒ = ℒℒNextbestmodel − ℒℒC = −6806 when we condition on the Vindija Neanderthal genome and Δℒℒ = −6240 when we condition on the Denisovan genome; table S4 and Materials and Methods). Our analyses provide support for a contribution to the genetic ancestry of present-day West African populations from an archaic ghost population whose divergence from the ancestors of modern humans predates the split of Neanderthals and modern humans.
We applied approximate Bayesian computation (ABC) to the CSFS to refine the parameters of our most likely demographic model (model C) (section S5). Given the large number of parameters in this demographic model, we fixed parameters that had previously been estimated (15) and jointly estimated the split time of the introgressing archaic population from the ancestors of Neanderthals and modern humans, the time of introgression, the fraction of ancestry contributed by the introgressing population, and its effective population size. We determined the posterior mean for the split time to be 625,000 years before the present (B.P.) [95% highest posterior density interval (HPD): 360,000 to 975,000], the admixture time to be 43,000 years B.P. (95% HPD: 6000 to 124,000), and the admixture fraction to be 0.11 (95% HPD: 0.045 to 0.19). Analyses of three other West African populations (ESN, GWD, and MSL) yielded concordant estimates for these parameters (Fig. 2 and table S7). Combining our results across the West African populations, we estimate that the archaic population split from the ancestor of Neanderthals and modern humans 360 thousand years (ka) to 1.02 million years (Ma) B.P. and subsequently introgressed into the ancestors of present-day Africans 0 to 124 ka B.P. contributing 2 to 19% of their ancestry. We caution that the true underlying demographic model is likely to be more complex. To explore aspects of this complexity, we examined the possibility that the archaic population diverged at the same time as the split time of modern humans and Neanderthals and found that this model can also produce a U-shaped CSFS with a likelihood that is relatively high, although lower than that of our best-fit model (Δℒℒ = −2713 for the Neanderthal CSFS and Δℒℒ = −2597 for the Denisovan CSFS, KS P ≤ 2.9 × 10−6). Our estimates of a large effective population size in the introgressing lineage (posterior mean of 25,000; 95% HPD: 23,000 to 27,000) could indicate additional structure. We find that the Ne of the introgressing lineage in YRI and MSL is larger than that in the other African populations, possibly due to a differential contribution from a basal West African branch (20).

Fig. 2 ABC estimates of the demographic parameters of the archaic ghost population across four West African populations (YRI, ESN, GWD, and MSL).
Posterior means are denoted by diamonds, and 95% credible intervals are denoted by lines. (A) The admixture time ta, (B) the admixture fraction α, (C) the split time of the introgressing population ts, and (D) the effective population size of the introgressing population Ne are shown. The parameter estimates are largely consistent across the African populations: We estimate split times of 360 ka to 1.02 Ma B.P., admixture times of 0 to 124 ka B.P., admixture fractions that range from 0.02 to 0.19, and effective population sizes that range from 22,000 to 28,000.
While we have chosen to represent the genetic contribution of the African ghost population as a single discrete interbreeding event, a more realistic model could include low levels of gene flow in a structured population over an extended period of time. Previously proposed models of ancestral structure in Africa do not fit the CSFS [KS P < 2 × 10−16 for the model described in (21) and KS P < 2 × 10−16 for the model proposed in (14); fig. S18], although we observe that the model of ancestral structure proposed by Yang et al. does produce a slight U-shape. We explored additional models of population structure in Africa (22) in which a lineage split from the ancestor of the modern humans with split times ranging from 100 to 550 ka B.P. and continued to exchange genes with the modern human population until the present with migration rates ranging from 2.5 × 10−5 to 2 × 10−2 migrants per generation. While these models of continuous gene flow produce a U-shaped CSFS for low migration rates and deep splits, they do not provide an adequate fit to the empirical CSFS over the range of parameters considered (KS P ≤ 2.3 × 10−5; section S6 and figs. S14 and S15). We used our ABC framework to explore a more detailed model of continuous migration in which we varied split time, migration rate, and effective population size of the introgressing lineage. Simulations under the best fitting model produce a CSFS that does not adequately fit the data (KS P = 1.83 × 10−6). A possible reason why the continuous migration models that we have explored do not fit the data is that these models can be considered as extensions of model A with multiple admixture events. We have shown that these models can only produce symmetric CSFS, unlike the CSFS that we observe in the data (appendix B). Thus, deep population structure within Africa alone cannot not explain the data (section S6).
Given the uncertainty in our estimates of the time of introgression, we wondered whether jointly analyzing the CSFS from both the CEU (Utah residents with Northern and Western European ancestry) and YRI genomes could provide additional resolution. Under model C, we simulated introgression before and after the split between African and non-African populations and observed qualitative differences between the two models in the high-frequency–derived allele bins of the CSFS in African and non-African populations (fig. S40). Using ABC to jointly fit the high-frequency–derived allele bins of the CSFS in CEU and YRI (defined as greater than 50% frequency), we find that the lower limit on the 95% credible interval of the introgression time is older than the simulated split between CEU and YRI (2800 versus 2155 generations B.P.), indicating that at least part of the archaic lineages seen in the YRI are also shared with the CEU (section S9.2).
We then attempted to understand the fine-scale distribution of archaic ghost ancestry along the genomes of present-day Africans. We used a recently developed statistical method (ArchIE) that combines multiple population genetic statistics to identify segments of diverged ancestry in 50 YRI and 50 MSL genomes without the need for an archaic reference genome (section S7) (23). Briefly, the method uses summary statistics computed from present-day genome sequences as input to a logistic regression model to estimate the probability that a haploid segment of an individual genome (defined as a contiguous region of length 50 kilobases) is archaic. While the parameters of the model are estimated by simulating data under a model that closely matches the demographic history relating Neanderthals and non-Africans, we found that ArchIE has 68% power to detect archaic segments at a false discovery rate of about 7% under our best-fit demographic model, confirming that its inferences are robust and sensitive to archaic introgression in Africa.
On average, ≃6.6 and ≃7.0% of the genome sequences in YRI and MSL were labeled as putatively archaic in ancestry. We sought to test whether the putatively archaic segments identified in YRI and MSL traced their primary ancestry to other African populations (8–10) or to known archaic hominins such as the Neanderthals or Denisovans. We computed the divergence of these segments to a genome sequence from each of six populations: southern African KhoeSan, Jul‘hoan; two Central African pygmy populations (Biaka and Mbuti); and two archaic hominin populations (Neanderthal and Denisovan). We expect segments introgressed from any of these populations to be less diverged relative to nonarchaic segments. On the contrary, the putatively archaic segments are more diverged, consistent with their source not being any of these populations (Fig. 3C and section S7.1). Merging the putatively archaic segments across individual genomes, we obtained a total of 482 and 502 Mb of archaic genome sequence in the YRI and MSL, respectively. We estimated the distribution of the time to the most recent common ancestor (TMRCA) between segments labeled archaic and those labeled nonarchaic using the pairwise mode of multiple sequentially Markovian coalescent (MSMC) (Fig. 3B and section S7.2) (24) and observed that the TMRCA is larger for the putatively archaic class of segments. Specifically, we find that the median nonarchaic segment coalescent time is 0.865 Ma ago for both populations, while the median archaic segment coalescent time is 1.51 Ma ago for YRI and 1.15 Ma ago for MSL (1.69- and 1.23-fold increases in age for YRI and MSL, respectively).
|
|
|
|
Post by Admin on Feb 16, 2020 21:58:24 GMT
 Fig. 3 Analysis of segments of archaic ghost ancestry found in the Yoruba and Mende populations. (A) Inference of segments of archaic ancestry was performed with ArchIE. ArchIE proceeds by simulating data under a model of archaic introgression, calculating population genetic summary statistics, and training a model to predict the probability that a 50-kb window in an individual comes from an archaic population. We apply the resulting predictor to genome sequences from the Yoruba and Mende populations. (B) Comparison of TMRCA between inferred archaic and nonarchaic segments to the TMRCA of a pair of nonarchaic segments in the Yoruba. On average, archaic segments are 1.69× older than nonarchaic segments. (C) Estimates of the divergence times of archaic segments inferred in Yoruba from KhoeSan, Jul‘hoan, two modern human pygmy genomes (Mbuti and Biaka), and Neanderthal and Denisovan genomes compared to divergence times of nonarchaic segments. P values are computed via block jackknife. Archaic segments are more diverged from all six genomes than nonarchaic segments. We examined the frequencies of archaic segments to investigate whether natural selection could have shaped the distribution of archaic alleles (fig. S40). We found 33 loci with an archaic segment frequency of ≥50% in the YRI (a cutoff chosen to be larger than the 99.9th percentile of introgressed archaic allele frequencies based on a neutral simulation of archaic introgression with parameters related to the time of introgression and admixture fraction chosen conservatively to maximize the drift since introgression; section S7.3 and fig. S40) and 37 loci in the MSL. Some of these genes are at high frequency across both the YRI and MSL, including NF1, a tumor suppressor gene (83% in YRI, 85% in MSL), MTFR2, a gene involved with mitochondrial aerobic respiration in the testis (67% in YRI, 78% in MSL), HSD17B2, a gene involved with hormone regulation (74% in YRI, 68% in MSL), KCNIP4, which is a gene involved with potassium channels (73% in YRI, 69% in MSL), and TRPS1, a gene associated with trichorhinophalangeal syndrome (71% in YRI, 75% in MSL; Table 1). Three of these genes have been found in previous scans for positive selection in the YRI: NF1 (25, 26), KCNIP4 (27), and TRPS1 (28). On the other hand, we do not find elevated frequencies at MUC7, a gene previously found to harbor signatures of archaic introgression (29). DISCUSSION Our analyses document introgression in four present-day West African populations from an archaic population that likely diverged before the split of modern humans and the ancestors of Neanderthals and Denisovans. A number of previous studies have found evidence for deeply diverged lineages contributing genetic ancestry to the Pygmy (8, 9) and Yoruba (7, 30) populations. Analyses of ancient African genomes have revealed that stone-age hunter-gatherers from South Africa diverged from other modern-day populations >260,000 years (31) B.P. and that present-day West African populations trace part of their ancestry to a basal lineage that diverged before the split of the southern African San (20) (although an alternative model consistent with their data includes a complex pattern of isolation by distance between western, eastern, and southern African populations). Placing our results within the context of the complex patterns of deep divergences in the African populations will require the analysis of a diverse set of African populations that include the southern African San populations, as well as the inclusion of ancient African genomes that lack signals of recent admixture that are present in the present-day San populations (32). One interpretation of the recent time of introgression that we document is that archaic forms persisted in Africa until fairly recently (33). Alternately, the archaic population could have introgressed earlier into a modern human population, which then subsequently interbred with the ancestors of the populations that we have analyzed here. The models that we have explored here are not mutually exclusive, and it is plausible that the history of African populations includes genetic contributions from multiple divergent populations, as evidenced by the large effective population size associated with the introgressing archaic population. Relatively, recent fossils with archaic features (or combinations of archaic and modern human features) have been found in the fossil record in Africa and the Middle East. While anatomically modern humans appear in the fossil record around 200,000 years ago, fossils with a combination of archaic and modern features can be found across sub-Saharan Africa and the Middle East until as recently as 35,000 years ago (34). Examples of these fossils include a cranium from Iwo Eleru (33) and human remains from Ishango (35) that have been interpreted as being consistent with deep structure and representing a complex history of interaction between modern and archaic hominins in Africa. The signals of introgression in the West African populations that we have analyzed raise questions regarding the identity of the archaic hominin and its interactions with the modern human populations in Africa. Analysis of the CSFS in the Luhya from Webuye, Kenya (LWK) also reveals signals of archaic introgression, although our interpretation is complicated by recent admixture in the LWK that involves populations related to western Africans and eastern African hunter-gatherers (section S8) (20). Non-African populations (Han Chinese in Beijing and Utah residents with northern and western European ancestry) also show analogous patterns in the CSFS, suggesting that a component of archaic ancestry was shared before the split of African and non-African populations. A detailed understanding of archaic introgression and its role in adapting to diverse environmental conditions will require analysis of genomes from extant and ancient genomes across the geographic range of Africa. Science Advances 12 Feb 2020: Vol. 6, no. 7, eaax5097 |
|
|
|
Post by Admin on Feb 17, 2020 19:49:56 GMT
Abstract
A long-debated question concerns the fate of archaic forms of the genus Homo: did they go extinct without interbreeding with anatomically modern humans, or are their genes present in contemporary populations? This question is typically focused on the genetic contribution of archaic forms outside of Africa. Here we use DNA sequence data gathered from 61 noncoding autosomal regions in a sample of three sub-Saharan African populations (Mandenka, Biaka, and San) to test models of African archaic admixture. We use two complementary approximate-likelihood approaches and a model of human evolution that involves recent population structure, with and without gene flow from an archaic population. Extensive simulation results reject the null model of no admixture and allow us to infer that contemporary African populations contain a small proportion of genetic material (≈2%) that introgressed ≈35 kya from an archaic population that split from the ancestors of anatomically modern humans ≈700 kya. Three candidate regions showing deep haplotype divergence, unusual patterns of linkage disequilibrium, and small basal clade size are identified and the distributions of introgressive haplotypes surveyed in a sample of populations from across sub-Saharan Africa. One candidate locus with an unusual segment of DNA that extends for >31 kb on chromosome 4 seems to have introgressed into modern Africans from a now-extinct taxon that may have lived in central Africa. Taken together our results suggest that polymorphisms present in extant populations introgressed via relatively recent interbreeding with hominin forms that diverged from the ancestors of modern humans in the Lower-Middle Pleistocene.
H. sapienshybridization
It is now well accepted that anatomically modern humans (AMH) originated in Africa and eventually dispersed to all inhabited parts of the world. What is not known is the extent to which the ancestral population that gave rise to AMH was genetically isolated, and whether archaic hominins made a genetic contribution to the modern human gene pool. Answering these questions has important implications for understanding the way in which adaptations associated with modern traits were assembled in the human genome: do the genes of AMH descend exclusively from a single isolated population, or do our genes descend from divergent ancestors that occupied different ecological niches over a wider geographical range across and outside of the African Pleistocene landscape?
The introgression debate is typically framed in terms of interbreeding between AMH and Neandertals in Europe or other archaic forms in Asia. The opportunity for such hybridizations may have existed between 90 and 30 kya, after early modern humans dispersed from Africa and before archaic forms went extinct in Eurasia (1⇓⇓⇓–5). Recent genome-level analyses of ancient DNA suggest that a small amount of gene flow did occur from Neandertals into the ancestors of non-Africans sometime after AMH left Africa (6) and that an archaic “Denisovan” population contributed genetic material to the genomes of present-day Melanesians (7). Given recent fossil evidence, however, the greatest opportunity for introgression was in Africa, where AMH and various archaic forms coexisted for much longer than they did outside of Africa (5, 8–11). Indeed, the fossil record indicates that a variety of transitional forms with a mosaic of archaic and modern features lived over an extensive geographic area from Morocco to South Africa between 200 and 35 kya (12⇓⇓–15).
Although sequencing the Neandertal and Denisovan genomes has provided evidence that gene flow between archaic and modern humans is plausible, it has not aided efforts to determine the extent of introgression in African populations. Here we use a different strategy to address the question of ancient population structure in Africa. Using multilocus DNA sequence polymorphism data from extant Africans, we analyze patterns of divergence and linkage disequilibrium (LD) to detect the signature of archaic admixture (16⇓–18). Application of this approach to publicly available sequence data from the Environmental Genome Project found evidence of ancient population structure in both African and non-African populations (19). However, analyses of diversity in and around coding regions may be complicated by the effects of recent natural selection, which might contribute to unusual patterns of polymorphism. In this study we use a large resequencing dataset that includes 61 noncoding regions on the autosomes to test whether patterns of neutral polymorphism in three contemporary sub-Saharan African populations are better explained by archaic admixture. Although whole-genome polymorphism data are now available from hundreds of samples (20), they do not include individuals from African hunter-gatherer populations, which serve as important reservoirs of human genetic diversity. Our study includes two such populations (Biaka Pygmies and San), along with an agricultural population from West Africa (Mandenka). We use a model of historically isolated subpopulations (17, 21) to predict patterns of nucleotide variation expected as a consequence of no admixture (null hypothesis) vs. low levels of admixture (alternative hypothesis). We apply two complementary coalescent-based approaches, a two-population and a three-population model to test the null hypothesis, and then estimate three key parameters: the time of admixture (Ta), the ancestral split time (T0), and the admixture proportion (a).
|
|

