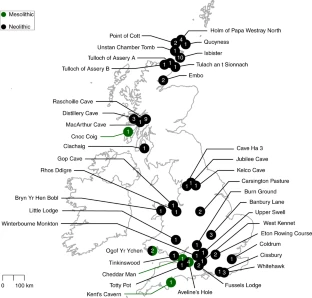
Post by Admin on Jan 13, 2020 21:03:32 GMT
Principal Component Analysis, f-Statistics, and Mixture Modeling
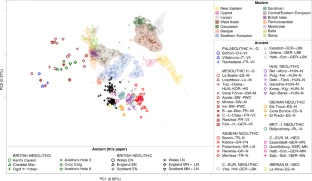
The first two dimensions of variation from principal component analysis (PCA) reveal a tight clustering of all five Aegean Neolithic genomes with Early Neolithic (EN) genomes from central and southern Europe (2, 3, 13) (Fig. 2). This cluster remains well-defined when the third dimension of variation is also considered (https://figshare.com/articles/Hofmanova_et_al_3D_figure_S4/3188767). Two recently published pre-Neolithic genomes from the Caucasus (20) appear to be highly differentiated from the genomes presented here and most likely represent a forager population distinct from the Epipaleolithic/Mesolithic precursors of the early Aegean farmers.

Fig. 2.
PCA of modern reference populations (18, 19) and projected ancient individuals. The Greek and Anatolian samples reported here cluster tightly with other European farmers close to modern-day Sardinians; however, they are clearly distinct from previously published Caucasian hunter-gatherers (20). This excludes the latter as a potential ancestral source population for early European farmers and suggests a strong genetic structure in hunter-gatherers of Southwest Asia. Central and East European (C./E. European), South European (South Eur.). Ancient DNA data: Pleistocene hunter-gatherer (Plei. HG) (20, 21, 22), Holocene hunter-gatherer (Holocene HG) (2, 4, 13, 20, 23), Neolithic (2, 4, 12, 13, 24), Late Neolithic/Chalcolithic/Copper Age (LN/Chalc./CA) (13, 25), and Bronze Age (13). Ancient samples are abbreviated consistently using the nomenclature “site-country code-culture”; see SI Appendix, Table S14 and Dataset S1 for more information. A 3D PCA plot can be viewed as a 3D figure (https://figshare.com/articles/Hofmanova_et_al_3D_figure_S4/3188767).
To examine this clustering of Early Neolithic farmers in more detail, we calculated outgroup f3 statistics (26) of the form f3 (‡Khomani; TEST, Greek/Anatolian), where TEST is one of the available ancient European genomes (SI Appendix, SI7. Using f-statistics to Infer Genetic Relatedness and Admixture Amongst Ancient and Contemporary Populations and Figs. S8–S10; Dataset S2); ‡Khomani San were selected as an outgroup as they are considered to be the most genetically diverged extant human population. Consistent with their PCA clustering, the northern Aegean genomes share high levels of genetic drift with each other and with all other previously characterized European Neolithic genomes, including early Neolithic from northern Spain, Hungary, and central Europe. Given the archaeological context of the different samples, the most parsimonious explanation for this shared drift is migration of early European farmers from the northern Aegean into and across Europe (12).
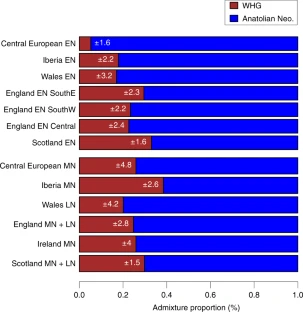
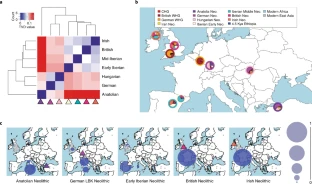
To better characterize this inferred migration, we modeled ancient and modern genomes as mixtures of DNA from other ancient and/or modern genomes, a flexible approach that characterizes the amount of ancestry sharing among multiple groups simultaneously (18, 27) (Fig. 3; SI Appendix, SI10. Comparing Allele Frequency Patterns Among Samples Using a Mixture Model). Briefly, we first represented each ancient or modern “target” group by the (weighted) number of alleles that they share in common with individuals from a fixed set of sampled populations (i.e., the “unlinked” approach described in ref. 27), which we refer to as the “allele-matching profile” for that target group. To cope with issues such as unequal sample sizes, we then used a linear model (28) to fit the allele-matching profile of the target group as a mixture of that of other sampled groups. Sampled groups that contribute most to this mixture indicate a high degree of shared ancestry with the target group relative to other groups. Under this framework the oldest Anatolian genome (Bar31) was inferred to contribute the highest amount of genetic ancestry (39–53%) to the Early Neolithic genomes from Hungary (13) and Germany (2) compared with any other ancient or modern samples, with the next highest contributors being other ancient Aegean genomes (Klei10, Pal7, Bar8) (SI Appendix, Figs. S23, S24, and S29). This pattern is not symmetric in that we infer smaller contributions from the German (<26%) and Hungarian (<43%) Neolithic genomes to any of the Anatolian or Greek ancient genomes. Furthermore, in this analysis modern samples from Europe and surrounding regions are inferred to be relatively more genetically related to the Aegean Neolithic genomes than to the Neolithic genomes from Germany and Hungary (Fig. 3; SI Appendix, SI10. Comparing Allele Frequency Patterns Among Samples Using a Mixture Model). These patterns are indicative of founder effects (29) in the German and possibly Hungarian Neolithic samples from a source that appears to be most genetically similar to the Aegean Neolithic samples (specifically, Bar31) and that distinguishes them from the ancestors of modern groups. Consistent with this, we found fewer short runs of homozygosity (ROH) (between 1 and 2 Mb) in our high-coverage Anatolian sample (Bar8) than in Early Neolithic genomes from Germany and Hungary (SI Appendix, SI11. Runs of Homozygosity and Fig. S31). However, it is not possible to infer a direction for dispersal within the Aegean with statistical confidence because both the Greek and Anatolian genomes copy from each other to a similar extent. We therefore see the origins of European farmers equally well represented by Early Neolithic Greek and northwestern Anatolian genomes.

Fig. 3.
Inferred mixture coefficients when forming each modern (small pies) and ancient (large pies, enclosed by borders matching key at left) group as a mixture of the modern-day Yoruba from Africa and the ancient samples shown in the key at left.
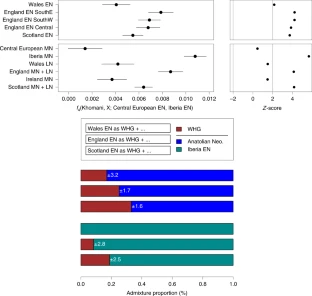
Ongoing gene flow into and across the Aegean is also indicated in the genome of a Chalcolithic individual from Kumtepe [Kum6 (25)], a site geographically close to Barcın but dating to ∼1,600 y later. Although archaeological evidence indicates a cultural break in many Aegean and West Anatolian settlements around 5,700/5,600 cal BCE [i.e., spanning this 1,600-y period (30)], Kum6 shows affinities to the Barcın genomes in “outgroup” f3-statistics in the form f3 (‡Khomani; TEST, Greek/Anatolian). The shared drift between Kum6 and both the early and late Neolithic Aegeans is similar in extent to the drift that Aegeans share with one another. However, f4 statistics of the form f4 (Aegean, Kum6, Early farmer, ‡Khomani) were often significantly positive (SI Appendix, Table S22; Dataset S2), suggesting that European Neolithic farmers [namely, Linearbandkeramik (LBK), Starcevo, and Early Hungarian Neolithic farmers] share some ancestry with early Neolithic Aegeans that is absent in Kum6. This is consistent with population structure in the Early Neolithic Aegean or with Kum6 being sampled from a population that differentiated from early Neolithic Aegeans after they expanded into the rest of Europe. Accordingly, compared with Barcın, Kum6 shares unique drift with the Late Neolithic genomes from Greece (Klei10 and Pal7), consistent with ongoing gene flow across the Aegean during the fifth millennium and with archaeological evidence demonstrating similarities in Kumtepe ceramic types with the Greek Late Neolithic (31). Finally, the Kum6, Klei10, and Pal7 genomes show signals of Caucasus hunter-gatherer (20) admixture that is absent in the Barcın genomes, suggesting post early Neolithic gene flow into the Aegean from the east.
It is widely believed that farming spread into Europe along both Mediterranean and central European routes, but the extent to which this process involved multiple dispersals from the Aegean has long been a matter of debate (32). We calculated f4 statistics to examine whether the Aegean Neolithic farmers shared drift with genomes from the Spanish Epicardial site Els Trocs in the Pyrenees (3, 12) that is distinct from that shared with Early Neolithic genomes from Germany and Hungary. In a test of the form f4 (Germany/Hungary EN, Spain EN, Aegean, ‡Khomani), we infer significant unique drift among Neolithic Aegeans (not significantly in Bar8) and Early Neolithic Spain to the exclusion of Hungarian and German Neolithic genomes (SI Appendix, Table S21). The best explanation for this observation is that migration to southwestern Europe started in the Aegean but was independent from the movement to Germany via Hungary. This is also supported by other genetic inferences (24) and archaeological evidence (33). An alternative scenario is a very rapid colonization along a single route with subsequent gene flow back to Greece from Spain. Potentially, preexisting hunter-gatherer networks along the western Mediterranean could have produced a similar pattern, but this is not supported by archaeological data. Interestingly, Ötzi the Tyrolean Iceman (11) shows unique shared drift with Aegeans to the exclusion of Hungarian Early Neolithic farmers and Late and Post Neolithic European genomes and feasibly represents a relict of Early Neolithic Aegeans (SI Appendix, SI7. Using f-statistics to Infer Genetic Relatedness and Admixture Amongst Ancient and Contemporary Populations and Table S18).
The first two dimensions of variation from principal component analysis (PCA) reveal a tight clustering of all five Aegean Neolithic genomes with Early Neolithic (EN) genomes from central and southern Europe (2, 3, 13) (Fig. 2). This cluster remains well-defined when the third dimension of variation is also considered (https://figshare.com/articles/Hofmanova_et_al_3D_figure_S4/3188767). Two recently published pre-Neolithic genomes from the Caucasus (20) appear to be highly differentiated from the genomes presented here and most likely represent a forager population distinct from the Epipaleolithic/Mesolithic precursors of the early Aegean farmers.
Fig. 2.
PCA of modern reference populations (18, 19) and projected ancient individuals. The Greek and Anatolian samples reported here cluster tightly with other European farmers close to modern-day Sardinians; however, they are clearly distinct from previously published Caucasian hunter-gatherers (20). This excludes the latter as a potential ancestral source population for early European farmers and suggests a strong genetic structure in hunter-gatherers of Southwest Asia. Central and East European (C./E. European), South European (South Eur.). Ancient DNA data: Pleistocene hunter-gatherer (Plei. HG) (20, 21, 22), Holocene hunter-gatherer (Holocene HG) (2, 4, 13, 20, 23), Neolithic (2, 4, 12, 13, 24), Late Neolithic/Chalcolithic/Copper Age (LN/Chalc./CA) (13, 25), and Bronze Age (13). Ancient samples are abbreviated consistently using the nomenclature “site-country code-culture”; see SI Appendix, Table S14 and Dataset S1 for more information. A 3D PCA plot can be viewed as a 3D figure (https://figshare.com/articles/Hofmanova_et_al_3D_figure_S4/3188767).
To examine this clustering of Early Neolithic farmers in more detail, we calculated outgroup f3 statistics (26) of the form f3 (‡Khomani; TEST, Greek/Anatolian), where TEST is one of the available ancient European genomes (SI Appendix, SI7. Using f-statistics to Infer Genetic Relatedness and Admixture Amongst Ancient and Contemporary Populations and Figs. S8–S10; Dataset S2); ‡Khomani San were selected as an outgroup as they are considered to be the most genetically diverged extant human population. Consistent with their PCA clustering, the northern Aegean genomes share high levels of genetic drift with each other and with all other previously characterized European Neolithic genomes, including early Neolithic from northern Spain, Hungary, and central Europe. Given the archaeological context of the different samples, the most parsimonious explanation for this shared drift is migration of early European farmers from the northern Aegean into and across Europe (12).
To better characterize this inferred migration, we modeled ancient and modern genomes as mixtures of DNA from other ancient and/or modern genomes, a flexible approach that characterizes the amount of ancestry sharing among multiple groups simultaneously (18, 27) (Fig. 3; SI Appendix, SI10. Comparing Allele Frequency Patterns Among Samples Using a Mixture Model). Briefly, we first represented each ancient or modern “target” group by the (weighted) number of alleles that they share in common with individuals from a fixed set of sampled populations (i.e., the “unlinked” approach described in ref. 27), which we refer to as the “allele-matching profile” for that target group. To cope with issues such as unequal sample sizes, we then used a linear model (28) to fit the allele-matching profile of the target group as a mixture of that of other sampled groups. Sampled groups that contribute most to this mixture indicate a high degree of shared ancestry with the target group relative to other groups. Under this framework the oldest Anatolian genome (Bar31) was inferred to contribute the highest amount of genetic ancestry (39–53%) to the Early Neolithic genomes from Hungary (13) and Germany (2) compared with any other ancient or modern samples, with the next highest contributors being other ancient Aegean genomes (Klei10, Pal7, Bar8) (SI Appendix, Figs. S23, S24, and S29). This pattern is not symmetric in that we infer smaller contributions from the German (<26%) and Hungarian (<43%) Neolithic genomes to any of the Anatolian or Greek ancient genomes. Furthermore, in this analysis modern samples from Europe and surrounding regions are inferred to be relatively more genetically related to the Aegean Neolithic genomes than to the Neolithic genomes from Germany and Hungary (Fig. 3; SI Appendix, SI10. Comparing Allele Frequency Patterns Among Samples Using a Mixture Model). These patterns are indicative of founder effects (29) in the German and possibly Hungarian Neolithic samples from a source that appears to be most genetically similar to the Aegean Neolithic samples (specifically, Bar31) and that distinguishes them from the ancestors of modern groups. Consistent with this, we found fewer short runs of homozygosity (ROH) (between 1 and 2 Mb) in our high-coverage Anatolian sample (Bar8) than in Early Neolithic genomes from Germany and Hungary (SI Appendix, SI11. Runs of Homozygosity and Fig. S31). However, it is not possible to infer a direction for dispersal within the Aegean with statistical confidence because both the Greek and Anatolian genomes copy from each other to a similar extent. We therefore see the origins of European farmers equally well represented by Early Neolithic Greek and northwestern Anatolian genomes.
Fig. 3.
Inferred mixture coefficients when forming each modern (small pies) and ancient (large pies, enclosed by borders matching key at left) group as a mixture of the modern-day Yoruba from Africa and the ancient samples shown in the key at left.
Ongoing gene flow into and across the Aegean is also indicated in the genome of a Chalcolithic individual from Kumtepe [Kum6 (25)], a site geographically close to Barcın but dating to ∼1,600 y later. Although archaeological evidence indicates a cultural break in many Aegean and West Anatolian settlements around 5,700/5,600 cal BCE [i.e., spanning this 1,600-y period (30)], Kum6 shows affinities to the Barcın genomes in “outgroup” f3-statistics in the form f3 (‡Khomani; TEST, Greek/Anatolian). The shared drift between Kum6 and both the early and late Neolithic Aegeans is similar in extent to the drift that Aegeans share with one another. However, f4 statistics of the form f4 (Aegean, Kum6, Early farmer, ‡Khomani) were often significantly positive (SI Appendix, Table S22; Dataset S2), suggesting that European Neolithic farmers [namely, Linearbandkeramik (LBK), Starcevo, and Early Hungarian Neolithic farmers] share some ancestry with early Neolithic Aegeans that is absent in Kum6. This is consistent with population structure in the Early Neolithic Aegean or with Kum6 being sampled from a population that differentiated from early Neolithic Aegeans after they expanded into the rest of Europe. Accordingly, compared with Barcın, Kum6 shares unique drift with the Late Neolithic genomes from Greece (Klei10 and Pal7), consistent with ongoing gene flow across the Aegean during the fifth millennium and with archaeological evidence demonstrating similarities in Kumtepe ceramic types with the Greek Late Neolithic (31). Finally, the Kum6, Klei10, and Pal7 genomes show signals of Caucasus hunter-gatherer (20) admixture that is absent in the Barcın genomes, suggesting post early Neolithic gene flow into the Aegean from the east.
It is widely believed that farming spread into Europe along both Mediterranean and central European routes, but the extent to which this process involved multiple dispersals from the Aegean has long been a matter of debate (32). We calculated f4 statistics to examine whether the Aegean Neolithic farmers shared drift with genomes from the Spanish Epicardial site Els Trocs in the Pyrenees (3, 12) that is distinct from that shared with Early Neolithic genomes from Germany and Hungary. In a test of the form f4 (Germany/Hungary EN, Spain EN, Aegean, ‡Khomani), we infer significant unique drift among Neolithic Aegeans (not significantly in Bar8) and Early Neolithic Spain to the exclusion of Hungarian and German Neolithic genomes (SI Appendix, Table S21). The best explanation for this observation is that migration to southwestern Europe started in the Aegean but was independent from the movement to Germany via Hungary. This is also supported by other genetic inferences (24) and archaeological evidence (33). An alternative scenario is a very rapid colonization along a single route with subsequent gene flow back to Greece from Spain. Potentially, preexisting hunter-gatherer networks along the western Mediterranean could have produced a similar pattern, but this is not supported by archaeological data. Interestingly, Ötzi the Tyrolean Iceman (11) shows unique shared drift with Aegeans to the exclusion of Hungarian Early Neolithic farmers and Late and Post Neolithic European genomes and feasibly represents a relict of Early Neolithic Aegeans (SI Appendix, SI7. Using f-statistics to Infer Genetic Relatedness and Admixture Amongst Ancient and Contemporary Populations and Table S18).