|
|
Post by Admin on Mar 15, 2014 0:01:49 GMT
It has been reported that the habitats of Neanderthals and ancestors of contemporary Eurasians overlapped both in time and space, and therefore provides possibility of introgression between Neanderthals and ancestors of Eurasians. This possibility is confirmed by recent studies, which suggest that about 1-4% of Eurasian genomes are from Neanderthal introgression. 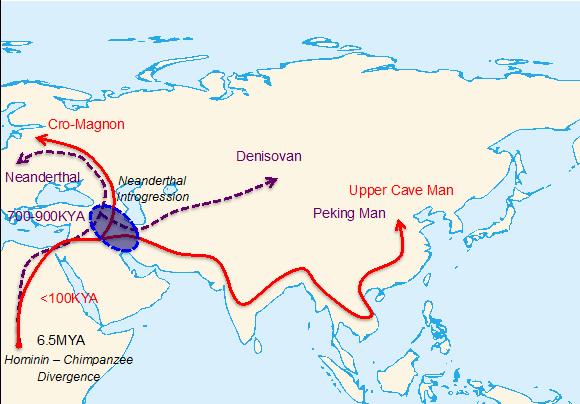 Two major out-of-Africa migration waves of hominins. The purple and red colors represent the first and second migration waves, respectively. The circle near Middle East represents a possible location where main Neanderthal introgression might have occurred. Adaptation to local environment is crucial for newly-arrived migrants, and the process of local adaptation is sometimes time-consuming. Since Neanderthals arrived in Eurasia ten times earlier than ancestors of Eurasians, we are trying to figure out whether the Neanderthal introgressions helped the ancestors of Eurasians adapt to the local environment. Our study reports that Neanderthal introgressive segments on chromosome 3 may have helped East Asians adapting to the intensity of ultraviolet-B (UV-B) irradiation in sunlight. We call the region containing the Neanderthal introgression the “HYAL” region, as it contains three genes that encode hyaluronoglucosaminidases. We first noticed that the entire HYAL region is included in an unusually large linkage disequilibrium (LD) block in East Asian populations. Such a large LD block is a typical signature of positive natural selection. More interestingly, it is observed that some Eurasian haplotypes at the HYAL region show a closer relationship to the Neanderthal haplotype than to the contemporary African haplotypes, implicating recent Neanderthal introgression. We confirmed the Neanderthal introgression in HYAL region by employing a series of statistical and population genetic analyses. Further, we examined whether the HYAL region was under positive natural selection using two published statistical tests. Both suggest that the HYAL region was under positive natural selection, and pinpoint a set of single nucleotide polymorphisms (SNPs) contributed by Neanderthal introgression as the candidate targets of positive natural selection. We then explored the potential functional importance of Neanderthal introgression in the HYAL region. The HYAL genes attracted our attention, as they are important in hyaluronan metabolism and cellular response to UV-B irradiation. We noticed that an SNP pinpointed as a potential target for positive natural selection was located in the most conservative exon of HYAL2 gene. We suspect that this SNP (known as rs35455589) may have altered the function of HYAL2 protein, since this SNP is also associated with the risk of keloid, a dermatological disorder related to hyaluronan metabolism.  Next, we interrogated the global distribution of Neanderthal introgression at the HYAL region. It is observed that the Neanderthal introgression reaches a very high frequency in East Asian populations, which ranges from 49.4% in Japanese to 66.5% in Southern Han Chinese. The frequency of Neanderthal introgression is higher in southern East Asian populations compared to northern East Asian populations. Such evidence might suggest latitude-dependent selection, which implicates the role of UV-B intensity. We discovered that Neanderthal introgression on chromosome 3 was under positive natural selection in East Asians. We also found that a gene (HYAL2) in the introgressive region is related to the cellular response to UV-B, and an allele from Neanderthal introgression may have altered the function of HYAL2. As such, we think it is possible that Neanderthals may have helped East Asians to adapt to sunlight. |
|
|
|
Post by Admin on Mar 28, 2014 6:10:30 GMT
 The 24,000-year-old remains of a young boy from the Siberian village of Mal’ta have added a new root to the family tree of indigenous Americans. While some of the New World's native ancestry clearly traces back to east Asia, the Mal’ta boy’s genome — the oldest known of any modern human — shows that up to one-third of that ancestry can be traced back to Europe. The results show that people related to western Eurasians had spread further east than anyone had suspected, and lived in Siberia during the coldest parts of the last Ice Age. “At some point in the past, a branch of east Asians and a branch of western Eurasians met each other and had sex a lot,” says palaeogeneticist Eske Willerslev at the University of Copenhagen, who led the sequencing of the boy’s genome. This mixing, he says, created Native Americans — in the sense of the populations of both North and South America that predated — as we know them. His team's results are published today in Nature1.  Two Khanty women in Man uskve nomad camp, Berezovsky, Khanty-Mansia, Russia The team found that DNA from the boy's mitochondria — the energy-processing organelles of living cells — belonged to a lineage called haplogroup U, which is found in Europe and west Asia but not in east Asia, where his body was unearthed. The result was so bizarre that Willerslev assumed that his sample had been contaminated with other genetic material, and put the project on hold for a year. But the boy’s nuclear DNA — the bulk of his genome — told the same story. “Genetically, this individual had no east Asian resemblance but looked like Europeans and people from west Asia,” says Willerslev. “But the thing that was really mind-blowing was that there were signatures you only see in today’s Native Americans.” This signal is consistent among peoples from across the Americas, implying that it could not have come from European settlers who arrived after Christopher Columbus. Instead, it must reflect an ancient ancestry. The Mal’ta boy’s genome showed that Native Americans can trace 14% to 38% of their ancestry back to western Eurasia, the authors conclude. Willerslev’s team suggests that after the ancestors of Native Americans split off from those of east Asians, they moved north. Somewhere in Siberia, they met another group of people coming east from western Eurasia — the people to whom the Mal’ta boy belonged. The two groups mingled, and their descendants eventually travelled east into North America. This new origin story helps to resolve several peculiarities in New World archaeology. For example, ancient skulls found in both North and South America have features that do not resemble those of East Asians. They also carry the mitochondrial haplogroup X, which is related to western Eurasian lineages but not to east Asian ones. On the basis of these features, some scientists have suggested that Native Americans descended from Europeans who sailed west across the Atlantic. However, says Willerslev, “you don’t need a hypothesis that extreme”. These features make sense when you consider that Native Americans have some western Eurasian roots.  Figure 2: Admixture graph for MA-1 and 16 complete genomes. Figure 2: Admixture graph for MA-1 and 16 complete genomes.We reconstructed admixture graphs using TreeMix21 to relate the population history of MA-1 to 11 modern genomes from worldwide populations22, 4 new genomes from Eurasia (Mari, Avar, Indian and Tajik ancestry) and the Denisova genome22 (Supplementary Information, section 11). The maximum-likelihood population tree inferred without admixture events places MA-1 on a branch that is basal to western Eurasians (Supplementary Fig. 12). However, a significant residual was observed between the empirical covariance for MA-1 and Karitiana, a Native American population, and the covariance predicted by the tree model (Supplementary Fig. 12). Consequently, gene flow between these lineages was inferred in all graphs incorporating two or more migration events (Fig. 2 and Supplementary Fig. 13). Bootstrap support for the migration edge from MA-1 to Karitiana, rather than from Karitiana to MA-1, was 99% in this analysis. We investigated further the population history of MA-1 by conducting sequence read-based D-statistic tests23 on proposed tree-like histories comprising MA-1 and combinations of 11 modern genomes (Supplementary Information, section 13). In agreement with the TreeMix results, these tests reject the tree ((X, Han), MA-1) where X represents Avar, French, Indian, Mari, Sardinian and Tajik, consistent with the MA-1 lineage sharing more recent ancestry with the western Eurasian branch after the split of Europeans and east Asians (Supplementary Table 13). This result also holds true when the Han Chinese is replaced with Dai, another east Asian population (Supplementary Table 13). Notably, we can also reject the tree ((Han,Karitiana),MA-1) (Z510.8), suggesting gene flow between MA-1 and ancestral Native Americans, in accordance with the admixture graphs (Supplementary Table 13). This result is consistent with allele frequency-based D-statistic tests20 on SNP arrays for 48 Native American populations of entirely First American ancestry19, indicating that all tested populations are equally related to MA-1 and that the admixture event occurred before the population diversification of the First American gene pool/ The genetic affinity between Native Americans and MA-1 could be explained by gene flow after the split between east Asians and Native Americans, either from theMA-1 lineage into Native American ancestors or from Native American ancestors to the ancestors of MA-1. However, MA-1, at approximately 24,000 cal. BP, pre-dates time estimates of the Native American–east Asian population divergence event24,25. This presents little time for the formation of a diverged Native American gene pool that could have contributed ancestry to MA-1, suggesting gene flow from the MA-1 lineage into Native American ancestors. Such gene flows hould also be detectable using modern-day western Eurasian populations in place of MA-1. Consistent with this, D-statistic tests western Eurasian ancestry and 62–86% east Asian ancestry, but we caution that these estimates assume unadmixed ancestral populations. Our study has four important implications. First, we find evidence that contemporary Native Americans and western Eurasians share ancestry through gene flow from a Siberian Upper Palaeolithic population into First Americans. Second, our findings may provide an explanation for the presence of mtDNA haplogroup X in Native Americans, which is related to western Eurasians but not found in east Asian populations29. Third, such an easterly presence in Asia of a population related to contemporary western Eurasians provides a possibility that non-east Asian cranial characteristics of the First Americans13 derived from the Old World via migration through Beringia, rather than by a trans-Atlantic voyage from Iberia as proposed by the Solutrean hypothesis30. Fourth, the presence of an ancient western Eurasian genomic signature in the Baikal area before and after the LGM suggests that parts of south-central Siberia were occupied by humans throughout the coldest stages of the last ice age. Raghavan, M. et al. Nature dx.doi.org/10.1038/nature12736 (2013). |
|
|
|
Post by Admin on Apr 10, 2014 15:34:12 GMT
The genetic similarities between certain human populations and Neanderthals are striking. Indeed, many researchers think the Europeans and Asians inherited between 1 and 4 percent of their DNA from Neanderthals, yet scientists have struggled to demonstrate with a high degree of certainty that these genetic similarities are the result of interbreeding between these two species. Now, a pair of European scientists say that they have confirmed the human-Neanderthal reproduction hypothesis using statistical modeling — and these results, the researchers add, should go a long way to change the way we think of other human-like species.  In the past, genetic similarities between Neanderthals and humans have been associated with two possible scenarios. The first hypothesis puts forth that idea that certain human populations — those that went on to become modern Eurasians — evolved in isolated patches in Africa that allowed them to stay genetically similar to Neanderthals after they split from their shared common ancestor. The interbreeding hypothesis, on the other hand, states that bouts of human-Neanderthal reproduction would have occurred after humans migrated out of Africa. So, to find out which hypothesis fit humanity's genetic history more closely, the scientists tested the two hypotheses using a statistics and an evolutionary model. "We did a bunch of math to compute the likelihood of two different scenarios," says Laurent Frantz, study co-author and evolutionary biologist at Wageningen University in the Netherlands. "We were able to do that by dividing the genome in small blocks of equal lengths from which we inferred genealogy." This method allowed the researchers to support with a high degree of certainty that interbreeding occurred, Frantz says. "Our analysis shows that a model that involves interbreeding is much more likely than a model where there was sustained substructure in Africa." The scientist cautions that sustained substructure might still have occurred, "but it cannot be used to explain the genetic similarities" all on its own.  These results, published today in Genetics, go against a 2012 study, published in Proceedings of the National Academy of Sciences, which found that interbreeding was far less likely than the alternative. "There seemed to be something that has gone wrong [in that study] because it seems unparsimonious to me," Frantz says. "When we tested two hypotheses, we got a high support for a scenario where humans and Neanderthals interbred." The researchers originally developed the statistical method to study the genetic history of insect and pig populations in Europe and Southeast Asia, respectively. But they think that it can also be used to study interbreeding events when there is a limited pool of genetic samples available. Furthermore, Frantz thinks that these results, along with those from previous studies, should serve to shift the conversation away from the brutality of human evolution. "There have been a lot of arguments about what happened to these species," the researcher says. "Some think that we outcompeted [other hominins] or that they were killed by humans, but now we can see that it's not that simple." In all likelihood, some Neanderthals were recruited into certain human populations, he says, and shared in their daily lives. So thinking of humanity solely in terms of a struggle to destroy all that differs from our species is, at least partially, incorrect. There is little doubt now, Frantz says, that "human evolution is much more complex than we previously thought." Lohse, Konrad, and Laurent AF Frantz. " Neandertal Admixture in Eurasia Confirmed by Maximum Likelihood Analysis of Three Genomes." Genetics (2014): genetics-114. |
|
|
|
Post by Admin on Apr 17, 2014 13:36:02 GMT
The study (Canfield et al. 2013) shows that Europeans and East Asians share common ancestry and they split around 50,000 years ago with a lighter skin mutation. It's known that both Europeans (5.9±0.08%) and East Asians (6.2±0.06%) inherited some portions of Neanderthal DNA because human-Neanderthal admixture occurred somewhere in the Middle East prior to Europeans' genetic split from proto-Asians, while Africans had not interbred with Neanderthals. Environmental factors may have contributed to the further differentiation of the two ethnic groups after the A111T mutation, which gave rise to modern Caucasians with lighter skin tones. Europeans who lived in colder climates in Scandinavia were positively selected to accumulate fat more effectively than East Asians to cope better with the cold weather, while East Asians went through another genetic mutation to adapt to the humid environment in Asia. This process of positive selection may explain the physical differences between Europeans and East Asians and Europeans are prone to gain more weight than their Asian counterparts, who are rarely overweight without making conscious efforts to lose weight. In other words, the metabolite concentration associated with Neanderthal ancestry among contemporary Europeans is directly linked to obesity, hypertriglyceridemia and coronary heart disease and Europeans are three times more likely to have a body mass index (BMI) of more than 40 (class III obesity) than Asians. 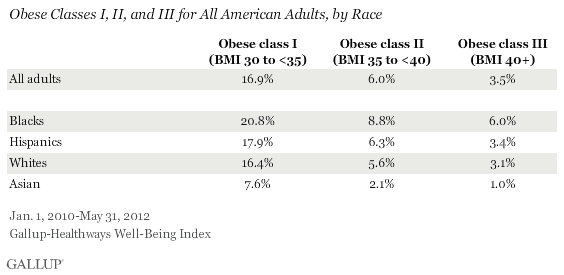 Although Neanderthals are extinct, fragments of their genomes persist in contemporary humans. Here we show that while the genome-wide frequency of Neanderthal-like sites is approximately constant across all contemporary out-of-Africa populations, genes involved in lipid catabolism contain more than threefold excess of such sites in contemporary humans of European descent. Evolutionally, these genes show significant association with signatures of recent positive selection in the contemporary European, but not Asian or African populations. Functionally, the excess of Neanderthal-like sites in lipid catabolism genes can be linked with a greater divergence of lipid concentrations and enzyme expression levels within this pathway, seen in contemporary Europeans, but not in the other populations. We conclude that sequence variants that evolved in Neanderthals may have given a selective advantage to anatomically modern humans that settled in the same geographical areas. The ancestors of Neanderthals and modern humans diverged from a common ancestral population ~800–400 thousand years ago (KYA)1, 2, 3, 4, 5. Subsequently, Neanderthals evolved in Europe and Central Asia and possibly spread further east into the Asian continent6. The first successful migration of modern humans out of Africa can be traced back in the archaeological and genetic records to ~70–60 KYA7. After that, modern humans spread quickly across all continents and could have coexisted with Neanderthals in Europe and Central Asia for thousands of years before the Neanderthal extinction 45–30 KYA7, 8. Studies comparing the complete nuclear genome sequences of Neanderthals and contemporary modern humans indicate that out-of-Africa human populations, but not sub-Saharan African populations, contain genomic regions with unusually high similarity to the Neanderthal genome9, 10. In each individual, these regions were reported to occupy 1–4% of the total genome sequence10. This phenomenon implies the occurrence of gene flow from Neanderthals into those human populations that migrated from the African continent11, 12.  Figure 1: Proportions of NLS in contemporary human populations. In this study, we asked whether genomic regions with high sequence similarity to the Neanderthal genome are distributed randomly across the genomes of contemporary modern humans. If certain regions in the modern human genome show a decreased Neanderthal gene flow, these regions may contain genetic changes essential to the modern human phenotype that are undergoing purifying selection against the Neanderthal alleles. By contrast, the presence of genomic regions experiencing excessive gene flow from Neanderthal may indicate that genetic changes that evolved in Neanderthals gave modern humans carrying the Neanderthal genotype a selective advantage. In agreement with previous observations10, the genomes of contemporary humans of European and Asian descent showed greater similarities to the Neanderthal genome than did the genomes of the three populations of purely African descent. On average, the NLS frequency was 6.1±0.2% for contemporary humans of European and Asian descent, thus indicating a substantial excess of NLS in contemporary out-of-Africa populations (Fig. 1b,c, blue bars; Supplementary Tables 1–3). This D-statistic estimate is similar to the ones reported by other studies (4.8±0.2%)10, with the higher values obtained in our study potentially arising from the additional filtering of genomic sites polymorphic in Neanderthals. Further, in agreement with other studies10, there was no substantial difference in the genome-wide frequencies of NLS between European and Asian populations, with a slight tendency for higher frequencies in Asians: 5.9±0.08 and 6.2±0.06%, respectively18, 19.  Figure 2: Outstanding genetic features of lipid catabolism genes. The excess of NLS observed in LCP genes for populations of European descent was based on a large number of sites (n=498), and was robust to bootstrapping across sites (P<0.01, 1,000 bootstraps, Supplementary Table 5; Supplementary Fig. 2). Notably, NLS were located in 23 independent genomic regions. Among the remaining 15 LCP genes that did not contain NLS, 8 did not contain sites showing divergence between Neanderthals and chimpanzees and the remaining 7 contained only a few such divergent sites (Supplementary Table 6). It is furthermore robust to the potential effects of DNA damage characteristic of ancient DNA samples, as excluding the C/T and A/G substitutions that may stem from deamination of cytosine residues in ancient DNA22, 23 did not affect the results (Supplementary Table 4). Repeating the analysis using the genome sequences of African (ASW, LWK, YRI), European (CEU, FIN, GBR, IBS) and East Asian (CHB, JPT) individuals, which were sequenced to deeper coverage at the pilot stage of the 1,000 genomes project, as well as the high-coverage genome sequences of African (ASW, LWK, YRI, MKK—Maasai in Kinyawa, Kenya), European (CEU, TSI) and East Asian (CHB, JPT) individuals provided by the Complete Genomics human diversity set24, confirmed our observations (Supplementary Tables 7 and 8). Finally, repeating the analysis using the high-coverage (Altai) and the low-coverage (Vindjia) Neanderthal genomes, separately, resulted in similar findings (Supplementary Table 9). The excess of NLS in LCP genes in the genomes of contemporary Europeans may be due to a rapid spread of Neanderthal alleles in European ancestors because of their adaptive significance. Specifically, one may hypothesize that, over time, Neanderthals acquired changes to lipid catabolism, which were beneficial for survival in the environmental conditions of prehistoric Europe and Central Asia. These adaptive variants may then have been acquired by the modern humans through introgression and rapidly brought to high frequency by positive selection. To test this hypothesis, we searched for signatures of positive selection in the genomes of contemporary humans of European, Asian and African decent using composite of multiple signals (CMS) scores25. High CMS values indicate genomic regions under recent positive selection based on three distinct signatures of selection: long-range haplotypes, differentiated alleles and high-frequency-derived alleles. We indeed found a significant excess of high CMS scores in the LCP gene regions of contemporary Europeans but not Asians or Africans (Fig. 2b). This effect was robust at different CMS score cutoffs and was specific to LCP: no significant excess of high CMS scores in individuals of European descent was observed in comparable genomic regions containing other metabolic genes (Supplementary Fig. 3). Furthermore, within the LCP term, high CMS scores found in contemporary Europeans were associated with genes containing the excess of NLS, but were not associated with other LCP genes (two-sample Wilcoxon test, P=0.0003; n=45 and 20; It is appealing to speculate that genetic variants affecting lipid catabolism in modern Europeans were acquired by modern human ancestors through genetic flow from Neanderthals, and then spread rapidly though the ancestral population by means of positive selection. Action of positive selection could be further explained by the adaptive significance of these variants in the geographic environment where Neanderthals had evolved and where Neanderthals and archaic Europeans later coexisted. It is noteworthy, however, that our observations are compatible with both introgression and incomplete lineage sorting hypotheses explaining the excess of genetic variants shared between contemporary humans and Neanderthals in out-of-Africa populations. Furthermore, while the excess of NLS in lipid catabolism genes is not observed in East Asian populations, we cannot assume that it is specific to Europeans. Complete genome sequences from a larger spectrum of human populations, especially from geographical regions coinciding with the Neanderthal living range, are needed to determine the full geographical spectrum of this effect. This is particularly true, given the fact that the Neanderthal area included Asian regions, such as the Altai Mountains. Still, the absence of NLS frequency increase in lipid catabolism genes in East Asian populations accompanied by no increase in the lipid concentration and enzyme expression divergence in the corresponding pathways indicates the geographical specificity of this phenomenon. One further argument indirectly supporting geographical specificity and local adaptive significance of the lipid catabolism changes potentially induced by Neanderthal variants comes from a comparison with the genome of another archaic human species—Denisovans36. Analysis of lipid catabolism gene sequences with the Denisova genome revealed a higher frequency of sites shared between Neanderthals and moderns humans, derived respective to both the chimpanzees and Denisovans, in contemporary Europeans than in East Asians (Supplementary Table 14). Notably, no such difference was observed in the genome-wide analysis. This result shows that changes in lipid catabolism genes shared between Neanderthals and contemporary Europeans were not fully present in Denisovans. Given that the presumed geographical area of Denisovans includes most of Asia36, this result indirectly supports specificity of observed lipid catabolism change to the European part of the Eurasian continent. We further note that a high frequency of NLS in lipid catabolism genes of contemporary Europeans does not require introgression, but is compatible with alternative scenarios. For instance, an alternative explanation of the general increase in NLS frequency in humans outside Africa, postulating the existence of a complex population structure within the African continent at the time of human and Neanderthal lineage divergence, has been hypothesized4. This hypothesis explains the presence of Neanderthal variants in non-African human populations by shared ancestry specific to ancestral human populations that left the African continent. If the lipid catabolism gene variants we find in Neanderthals and contemporary Europeans were already present in the ancestors of Neanderthals and out-of-Africa human populations, they may have independently increased in frequency in Neanderthals and humans situated in the European region. This scenario is probable if these genetic variants provided an adaptive advantage to both Neanderthal and human populations in the conditions of prehistoric Europe. While the presence of a recent positive selection signal in lipid catabolism gene variants containing NLS in modern Europeans supports such an adaptive scenario, the environmental pressures or functional mechanisms of this possible adaptive change remain elusive. Further studies of LCP conducted across multiple tissues and multiple contemporary human populations are needed to more fully assess the potential functional effects of this event. Khrameeva, Ekaterina E., et al. " Neanderthal ancestry drives evolution of lipid catabolism in contemporary Europeans." Nature communications 5 (2014). |
|
|
|
Post by Admin on Apr 30, 2014 4:51:18 GMT
Archaeologists have consistently speculated that the Basques moved into the region from southwestern Europe between 13,000 B.C. and 8,000 B.C. If this theory is correct, then it would be expected that both modern and ancient Basque DNA would be related to a specific DNA type originating in southwest Europe (which researchers term haplogroup V). In order to test this hypothesis, four genetic studies were performed on subjects from the modern Basque population during the 1990s. Utilizing mitochondrial DNA analysis (mtDNA) (Mitochondrial DNA Analysis - Modern Genetic Research Confirms Cayce's Story), geneticists found that the expected type of mtDNA (haplogroup V) occurred in 3.3 to 20 percent of modern Basques. In an effort to measure the mtDNA types present in the ancient Basques, a group of Spanish geneticists obtained dental samples from 121 individuals buried in four separate prehistoric Basque sites. In addition, bone samples (femurs) were used to confirm results. The burial sites were carbon dated from 3,000 B.C. to 1,400 B.C. The surprising results showed that not a single individual in the ancient group had the expected haplogroup V. The most frequent haplogroup found was H (at 37.2 percent). This type (H) is the most common mtDNA found in all modern-day European populations. In addition, 9.1 percent of the ancient Basque mtDNA was haplogroup X With the focus of their research on testing the hypothesis that haplogroup V moved into the Basque region in the years 13,000 B.C. to 8,000 B.C., the geneticists were forced to conclude that Haplogroup V entered the area after 3,000 B.C. They suggested that, prior to 9,000 B.C., various hunter-gatherer groups occupied the region. These groups included people from the X haplogroup. Extremely significant in light of the Cayce readings is the presence of haplogroup X in ancient Basque mtDNA. While popular press reports have often termed haplogroup X as "Caucasoid," this speculative idea has been generally discredited by researchers. In 1997, haplogroup X was discovered in about 3 percent of modern Native Americans and in ancient North American remains as well.  The X type is frequently found in modern descendants of the Iroquois and in ancient burials in Iroquois' lands. The X haplogroup has also been identified in the Middle East and, in 2001, it was found in a tribe living in the Altaic Mountains of the Gobi. All of these of course are areas where Cayce specifically stated Atlantean survivors fled in 10,000 B.C. The Editors of Ancient Mysteries, along with John Van Auken, have hypothesized that the X haplogroup may be the genetic link to the ancient Atlanteans. In books about Japan it is often remarked that many of the names of Japan's geographical features were taken over from the Ainu. For instance, the many names beginning or ending with ama (Goddess) are all thought to be of Ainu origin. In 1994 the newly married prince and princess of Japan traveled to the cave of the Goddess Amaterasu to ask her blessings for their marriage. The name Amaterasu is agglutinated from ama-atera-asu, ama (Goddess) atera (to come out, to appear) asturu (blessings flow): Blessings flow when the Goddess appears. This name is made up of perfect Basque! Other well-known names were similarly assembled such as Hokkaido: oka-aidu: oka (big meal) aiduru (looking forward to): Looking forward to a big meal; and Fujiyama, fa-uji-ama: fa (happy) uju (cry of joy) ama (Goddess): "A happy cry of joy for the Goddess" is uttered by everyone who reaches the top of the holy mountain, just like is still being heard on many other mountains of the world (e.g . at Croag Patrick in Ireland, on the last Sunday of July). The Basques even have a word for this yodel cry for the Goddess, which they call irrintzi. None of the Ainu words was the same as in Basque, but many were extremely close such as ikoro and koro (money), kokor and gogor (to scold), tasum and eritasun (illness), iska and xiska (to steal). A surprise was the Ainu word nok (testicle) which is much like the Basque word noka (familiarity with women). In English slang the same word is used in "to knock up" meaning "to cause a woman to become pregnant." In Indonesian nok means "unmarried young woman," while dénok means "slender, elegant woman." In Dutch slang the word is slightly altered to neuk (sexual intercourse). There is little doubt that the word goes way back to the Neolithic or even Paleolithic. From the following comparisons it seems clear that Ainu and Basque are genetically related. In comparing Ainu with Dravidian, there was no such a relationship, although Dravidian itself is obviously also related to Basque. Two separate branches of the same tree?  The Ainu language The Ainu language is genetically related to the universal language, Saharan/Basque; the similarities are just too many to be accidental. Considering that the Ainu have probably been separated from the west since 5-7,000 bce. it is not surprising that the language has drifted away from the Neolithic language as it had developed in the Sahara. The fact that so many Ainu words are still clearly recognizable when compared to modern Basque words is nothing short of amazing and tells us that the ancient oral traditions had been faithfully maintained since they left the Sahara or Mesopotamia. The Ainu had no writing system but memorized their history and legends as yukar, which means that the poetry and epics were performed by memory professionals with elaborate display and ritual. Similarly, in the west, the universal language was maintained by regular meetings, probably at the central shrine on Malta, where the bertsolari (memory professionals) of all the tribes and regions met to reinforce and standardize their language and knowledge. |
|

