|
|
Post by Admin on Jul 3, 2014 21:57:27 GMT
The origins of the First Americans remain contentious. Although Native Americans seem to be genetically most closely related to east Asians1, 2, 3, there is no consensus with regard to which specific Old World populations they are closest to4, 5, 6, 7, 8. Here we sequence the draft genome of an approximately 24,000-year-old individual (MA-1), from Mal’ta in south-central Siberia9, to an average depth of 1×. To our knowledge this is the oldest anatomically modern human genome reported to date. The MA-1 mitochondrial genome belongs to haplogroup U, which has also been found at high frequency among Upper Palaeolithic and Mesolithic European hunter-gatherers10, 11, 12, and the Y chromosome of MA-1 is basal to modern-day western Eurasians and near the root of most Native American lineages5. Similarly, we find autosomal evidence that MA-1 is basal to modern-day western Eurasians and genetically closely related to modern-day Native Americans, with no close affinity to east Asians. This suggests that populations related to contemporary western Eurasians had a more north-easterly distribution 24,000 years ago than commonly thought. Furthermore, we estimate that 14 to 38% of Native American ancestry may originate through gene flow from this ancient population. This is likely to have occurred after the divergence of Native American ancestors from east Asian ancestors, but before the diversification of Native American populations in the New World. Gene flow from the MA-1 lineage into Native American ancestors could explain why several crania from the First Americans have been reported as bearing morphological characteristics that do not resemble those of east Asians2, 13. Sequencing of another south-central Siberian, Afontova Gora-2 dating to approximately 17,000 years ago14, revealed similar autosomal genetic signatures as MA-1, suggesting that the region was continuously occupied by humans throughout the Last Glacial Maximum. Our findings reveal that western Eurasian genetic signatures in modern-day Native Americans derive not only from post-Columbian admixture, as commonly thought, but also from a mixed ancestry of the First Americans.  The genetic affinity between Native Americans and MA-1 could be explained by gene flow after the split between east Asians and Native Americans, either from theMA-1 lineage into Native American ancestors or from Native American ancestors to the ancestors of MA-1. However, MA-1, at approximately 24,000 cal. BP, pre-dates time estimates of the Native American–east Asian population divergence event24,25. This presents little time for the formation of a diverged Native American gene pool that could have contributed ancestry to MA-1, suggesting gene flow from theMA-1 lineage into NativeAmerican ancestors. Such gene flow should also be detectable using modern-daywestern Eurasian populations in place of MA-1. Consistent with this, D-statistic tests estimated from outgroup-ascertained SNP data20 reveal significant evidence (Z . 3) for Middle Eastern, European, central Asian and southAsian populations being closer toKaritiana than to HanChinese20 (Fig. 3b and Supplementary Information, section 14.5). Similar signals were also observed when we replaced modern-day Han Chinese with data from chromosome 21 from a 40,000-year-old east Asian individual (Tianyuan Cave, China), which has been found to be ancestral to modern-day Asians and Native Americans26 (Supplementary Information, section 14.5). Thus, if the gene flow direction was from Native Americans into western Eurasians it would have had to spread subsequently to European, Middle Eastern, south Asian and central Asian populations, including MA-1 before 24,000 years ago. Moreover, as Native Americans are closer to Han Chinese than to Papuans (Fig. 3c), NativeAmerican-related gene flowinto the ancestors ofMA-1 is expected to result in MA-1 also being closer to Han Chinese than to Papuans. However, our results suggest that this is not the case (D(Papuan, Han; Sardinian, MA-1)520.00260.005 (Z520.36)), which is compatible with all or almost all of the gene flow being into Native Americans (Supplementary Information, section 14.6).  Importantly, in addition to the low contamination rates and rare or extinct uniparental lineages, we exclude modern DNA contamination as being the source of the observed population affinities of MA-1 for three reasons. First, we corrected the sequence read-based D-statistics tests for differing amounts of contamination, using a European individual as the contamination source (Supplementary Information, section 13.5). We find similar outcomes for corrected and uncorrected tests (Supplementary Fig. 20), even when contamination levels larger than that estimated forMA-1 are considered, confirming that our results are not affected by contamination from a European source. Second, restricting the PCA to sequences with evidence of post-mortem degradation gives results that are comparable with those using the complete data set (Supplementary Information, section 15). Finally, the genome sequence of the researcher (Indian ancestry) who carried out DNA extraction and library preparation of MA-1 enables us to exclude the researcher as a source of contamination (Supplementary Information, sections 11 and 13). In addition, we exclude post-Columbian European admixture (after 1492 AD) as an explanation for the genetic affinity between MA-1 and Native Americans for three reasons. First, for SNP array-based analyses, we take recent European admixture into account by using a data set masked for inferred admixed genomic regions19. Second, allele frequency-based D-statistic tests20 show that all 48 tested modern-day populations with First American ancestry19 are equally related toMA-1within the resolution of our data (Supplementary Information, section 14.4), which would not be expected if the signal was driven by recent European admixture. Third, MA-1 is closer to Native Americans than any of the 15 tested European populations (Supplementary Information, section 14.8). 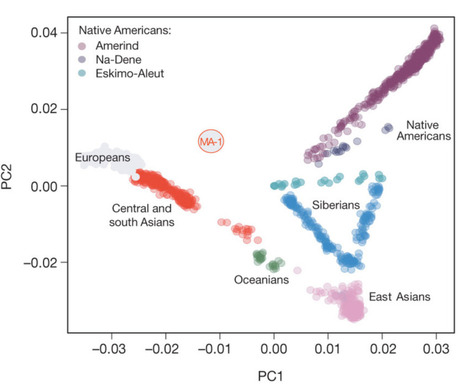 Human dispersals in northeast Asia immediately before and after the LGM are most likely to have led to the settlement of Beringia, and ultimately the Americas28. As MA-1 pre-dates the LGM, we investigated whether the genetic composition of southern Siberia changed during the LGM by generating a low-coverage data set (,0.13) of a post-LGM individual from Afontova Gora-2 (AG-2) (ref. 14), located on the western bank of the Enisei River in south-central Siberia (Fig. 1a). We obtained a direct AMS 14C date of 13,810 6 35 14C years before present or 17,075–16,750 cal. BP forAG-2 (Supplementary Information, section 2). Despite substantial present-day DNA contamination in this sample (Supplementary Information, section 5),wefind thatAG-2 shows close similarity to the genetic profile of MA-1 on a PCA (Supplementary Information, section 15 and Supplementary Fig. 29) and is significantly closer to Karitiana than to Han (D(Yoruba, AG-2; Han, Karitiana)50.07860.004, Z519.9) (Supplementary Information, section 15). We observe consistent results when restricting analyses to sequences with evidence of post-mortem degradation (Supplementary Information, section 15 and Supplementary Fig. 29), implying that southern Siberia may have experienced genetic continuity through the environmentally harsh LGM. Our study has four important implications. First, we find evidence that contemporary Native Americans and western Eurasians share ancestry through gene flow from a Siberian Upper Palaeolithic population into First Americans. Second, our findings may provide an explanation for the presence of mtDNA haplogroup X in Native Americans, which is related to western Eurasians but not found in east Asian populations29. Third, such an easterly presence in Asia of a population related to contemporary western Eurasians provides a possibility that non-east Asian cranial characteristics of the First Americans13 derived from the Old World via migration through Beringia, rather than by a trans-Atlantic voyage from Iberia as proposed by the Solutrean hypothesis30. Fourth, the presence of an ancient western Eurasian genomic signature in the Baikal area before and after theLGMsuggests that parts of south-central Siberia were occupied by humans throughout the coldest stages of the last ice age. Raghavan, Maanasa, et al. " Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans." Nature (2013). |
|
|
|
Post by Admin on Jul 7, 2014 0:47:00 GMT
The ancestry information provided by the 24 autosomal ASMs was first tested by performing a STRUCTURE analysis with the HGDP-CEPH samples assuming no prior knowledge of the ancestral groups. After K=4 the estimated loglikelihood of the data given the model (-19135) did not substantially change anymore. The four clusters detected at K=4 broadly match the four geographic regions: America, Sub-Saharan Africa, East Asia, and Eurasia (including Europe / Middle East / South Asia / Central Asia) (Figure 1). Only a small percentage of misclassified individuals was observed i.e., 0.47% Sub-Saharan Africans, 4.2% of Eurasians, 4.6% of Native American individuals, and 6.2% of East Asians (the latter was mainly in the Eurasian cluster with 3.6%). We concluded that these 24 SNPs are suitable for inferring bio-geographic ancestry in U.S. Americans since the four geographic regions identified represent the putative parental populations of the four major groups of U.S. Americans.  Figure 1. Genetic ancestry per individual in the global HGDP-CEPH panel as estimated by STRUCTURE using 24 autosomal ASMs (K=4). Figure 1. Genetic ancestry per individual in the global HGDP-CEPH panel as estimated by STRUCTURE using 24 autosomal ASMs (K=4).Next, we used the Native Americans, East Asians, Eurasians, and Sub-Sahara Africans from HGDP-CEPH as parental groups of the U.S. Americans (the genotype data of the 24 autosomal SNPs can be found in the Supp. Table S5) in a STRUCTURE analysis. Self-declared U.S. Europeans showed on average 93.2% of European ancestry (95% CI from 73.23% to 98.09%), self-declared U.S. Asians carried on average 89.5% of East Asian ancestry (95% CI from 37.43% to 97.46%), and self-declared U.S. Africans revealed on average 86.2 % Sub-Sahara African ancestry (95% CI from 47.82% to 98.5%) (Figure 2). For these three U.S. groups rather small (between 0.8 and 8.1% on average) components of continental ancestries other than the self-declared ones were detected (Figure 2). In contrast, self-declared U.S. Hispanics carried on average 61.2% European ancestry (95% CI from 8.33% to 95.75%), 14.9% Native American (95% CI from 1.21% to 55.54%), 10.8% East Asian (95% CI from 1.12% to 56.35%), and 11.6%, Sub-Saharan African ancestries (95% CI from 0.41% to 58.49%) (Figure 2). Furthermore, we observed for self-declared U.S. Africans statistically significant heterogeneity in the amount of African genetic ancestry depending on the geographic sampling region (Kruskal-Wallis test p-value=0.0042), as well as for self-declared U.S. Hispanics in the amount of Native American genetic ancestry (Kruskal-Wallis p-value = 1.48e-07). An AMOVA grouping individuals based on self-declared ancestry explained 34.2% (two tail p value <0.0005) of the total genetic variation suggesting strong genetic differentiation between self-declared ancestry groups of U.S. Americans.  Figure 2. Proportions of average continental genetic ancestry in four U.S. American groups of self-declared ancestry based on autosomal DNA, mtDNA and NRY DNA. Figure 2. Proportions of average continental genetic ancestry in four U.S. American groups of self-declared ancestry based on autosomal DNA, mtDNA and NRY DNA.From the MDS plot (Figure 3) it is evident that self-declared U.S. Europeans, U.S. Africans and U.S. Asians form rather discrete data clouds without strong overlaps between these groups, and tend to cluster close to their respective continental parental populations (from HGDP-CEPH). Self-declared U.S. Hispanics, however, did not cluster separately but either overlapped with U.S. / continental Europeans or appear between the U.S. / continental European cluster and the U.S. / continental Asian cluster with some U.S. Hispanics overlapping with the U.S. / continental African cluster or appeared between the U.S. / continental African and the U.S. / continental European clusters.  Figure 3. Two-dimensional plots of the first dimension, second dimension and third dimension obtained from a MDS analysis (stress = 0.13) performed with an Identical By State (IBS) distance matrix computed between pairs of individuals. Centroids of the four continental parental populations from HGDP-CEPH are marked by crosses. Figure 3. Two-dimensional plots of the first dimension, second dimension and third dimension obtained from a MDS analysis (stress = 0.13) performed with an Identical By State (IBS) distance matrix computed between pairs of individuals. Centroids of the four continental parental populations from HGDP-CEPH are marked by crosses.The current U.S. population represents a mixture of groups with different bio-geographic ancestries, mainly from Europe, Sub-Saharan Africa, East Asia and the Americas. We have shown in the HGPD-CEPH samples that the ascertained autosomal ASMs are informative for detecting the ancestry of these four continental groups. Overall, STRUCTURE, MDS and AMOVA analyses indicate that in U.S. Americans self-declared ancestry serves on average as a good proxy of the underlying autosomal genetic diversity, especially of European, African and Asian Americans. Our STRUCTURE results are in line with an earlier study reporting that ancestry self-identification corresponded well with STRUCTURE-based predictions for U.S. Americans (Tang, et al., 2005). Our findings with autosomal ASMs tend to corroborate previous findings performed in self-identified U.S. Europeans (Halder, et al., 2008; Halder, et al., 2009; Kosoy, et al., 2009) and U.S. Asians (Kosoy, et al., 2009), although usually many more markers were applied before. However, we observed discrepancies between our data and previous studies for self-declared U.S. Africans and U.S. Hispanics. For U.S. Africans we found a slightly larger percentage of African ancestry and a slightly lower percentage of European ancestry relative to previous reports (Tian, et al., 2006; Halder, et al., 2008; Halder, et al., 2009; Kosoy, et al., 2009; Zakharia, et al., 2009). For U.S. Hispanics, the Native American component tends to be rather low compared to previous studies (Price, et al., 2007; Halder, et al., 2009; Kosoy, et al., 2009). Differences in the admixture histories in different regions of the U.S. as reported elsewhere (Salazano and Bortolini, 2002; Kittles and Weiss, 2003; Zakharia, et al., 2009) are likely to explain such discrepancies. This view also is supported by the considerable heterogeneity in continental genetic ancestry depending on the geographic origin of the sampling region within the U.S. we observed for these two U.S. American groups. An alternative explanation in the case of U.S. Hispanics could be a lack of power of the set of autosomal ASMs we applied to distinguish Native American from East Asian ancestry (also explaining the apparent small Native American ancestry component in U.S. Asians). Native Americans and East Asians show a general genetic proximity due to their shared population history (Jakobsson, et al., 2008; Li, et al., 2008). Repeating the STRUCTURE analysis for U.S. Hispanics without considering East Asians as parental population raised the Native American ancestry component up to 27.44%, which is more comparable to previous studies. However, the fact that some of the self-declared U.S. Hispanic individuals carried NRY haplogroups typical for East Asians, and because a previous study also detected Asian ancestry in U.S. Hispanics (Guthery, et al., 2007), indicate that excluding East Asian admixture a priory would be incorrect for estimating genetic ancestry in U.S. Hispanics. Combining the ancestry information of patrilineal, matrilineal and biparental markers, a special quality of our study, offers the possibility to study the patterns of admixture at different levels of complexity. We observed the same degree of ancestry homogeneity in the three types of genetic markers for self-identified U.S. Europeans and U.S. Asians, which suggests relatively low genetic admixture with other ancestry groups than the one indicated by self-declaration. Noticeably, this finding for U.S. Europeans contrasts with common observation for self-declared European Americans from South America (Goncalves, et al., 2007; Corach, et al., 2010). In those South American groups European ancestry signals are usually high for NRY-DNA, intermediate for autosomal DNA, but low for mtDNA, whereas Native American genetic ancestry signals are reverse, indicating sex-bias admixture between mostly European men and mostly Native American women (Goncalves, et al., 2007; Corach, et al., 2010). This discrepancy between European Americans from North and South Americans has been explained in terms of local differences in social practices (Goncalves, et al., 2007). However, it could also be explained if the concept of ancestry self-identification had different meanings depending on the region of residence. This is supported by the fact that genetic admixture proportions of self-identified U.S. Hispanics from our study resemble those from self-declared European Americans in some South American countries with similar evidence for sex-biased admixture history. Our data also indicate sex-biased admixture for U.S. Africans with considerably more European NRY than mtDNA ancestry, and autosomal DNA estimates in-between. Previous studies analyzing NRY and mtDNA ancestry in U.S. Africans have reported similar results (Kayser, et al., 2003; Lind, et al., 2007), (see (Stefflova, et al., 2009) for a review), which we complement here with agreeing autosomal DNA evidence. Lao, Oscar, et al. " Evaluating self‐declared ancestry of US Americans with autosomal, Y‐chromosomal and mitochondrial DNA." Human mutation 31.12 (2010): E1875-E1893. |
|
|
|
Post by Admin on Jul 9, 2014 23:01:05 GMT
 Re-examination of a circa 100,000-year-old archaic early human skull found 35 years ago in Northern China has revealed the surprising presence of an inner-ear formation long thought to occur only in Neandertals. "The discovery places into question a whole suite of scenarios of later Pleistocene human population dispersals and interconnections based on tracing isolated anatomical or genetic features in fragmentary fossils," said study co-author Erik Trinkaus, PhD, a physical anthropology professor at Washington University in St. Louis."It suggests, instead, that the later phases of human evolution were more of a labyrinth of biology and peoples than simple lines on maps would suggest."  The study, forthcoming in the Proceedings of the National Academy of Sciences, is based on recent micro-CT scans revealing the interior configuration of a temporal bone in a fossilized human skull found during 1970s excavations at the Xujiayao site in China's Nihewan Basin. Trinkaus, the Mary Tileston Hemenway Professor in Arts & Sciences, is a leading authority on early human evolution and among the first to offer compelling evidence for interbreeding and gene transfer between Neandertals and modern human ancestors. His co-authors on this study are Xiu-Jie Wu, Wu Liu and Song Xing of the Institute of Vertebrate Paleontology and Paleoanthropology, Beijing, and Isabelle Crevecoeur of PACEA, Université de Bordeaux.  "We were completely surprised," Trinkaus said. "We fully expected the scan to reveal a temporal labyrinth that looked much like a modern human one, but what we saw was clearly typical of a Neandertal. This discovery places into question whether this arrangement of the semicircular canals is truly unique to the Neandertals."Often well-preserved in mammal skull fossils, the semicircular canals are remnants of a fluid-filled sensing system that helps humans maintain balance when they change their spatial orientations, such as when running, bending over or turning the head from side-to-side.  Since the mid-1990s, when early CT-scan research confirmed its existence, the presence of a particular arrangement of the semicircular canals in the temporal labyrinth has been considered enough to securely identify fossilized skull fragments as being from a Neandertal. This pattern is present in almost all of the known Neandertal labyrinths. It has been widely used as a marker to set them apart from both earlier and modern humans. The skull at the center of this study, known as Xujiayao 15, was found along with an assortment of other human teeth and bone fragments, all of which seemed to have characteristics typical of an early non-Neandertal form of late archaic humans. Trinkaus, who has studied Neandertal and early human fossils from around the globe, said this discovery only adds to the rich confusion of theories that attempt to explain human origins, migrations patterns and possible interbreedings. While it’s tempting to use the finding of a Neandertal-shaped labyrinth in an otherwise distinctly “non-Neandertal” sample as evidence of population contact (gene flow) between central and western Eurasian Neandertals and eastern archaic humans in China, Trinkaus and colleagues argue that broader implications of the Xujiayao discovery remain unclear. “The study of human evolution has always been messy, and these findings just make it all the messier,” Trinkaus said. “It shows that human populations in the real world don’t act in nice simple patterns. “Eastern Asia and Western Europe are a long way apart, and these migration patterns took thousands of years to play out,” he said. “This study shows that you can’t rely on one anatomical feature or one piece of DNA as the basis for sweeping assumptions about the migrations of hominid species from one place to another.” More information: Temporal labyrinths of eastern Eurasian Pleistocene humans, PNAS, www.pnas.org/cgi/doi/10.1073/pnas.1410735111Journal reference: Proceedings of the National Academy of Sciences |
|
|
|
Post by Admin on Jul 20, 2014 21:54:51 GMT
 The recent discovery of Denisovans (1,2) and genetic evidence of their hybridization with modern human populations now found in Island Southeast Asia, Australia, and the Pacific (3) are intriguing and unexpected. The reference specimen for the Denisovan genome(4), a distal phalanx from a young girl, was recovered fromthe geographically distant Denisova Cave in the Russian Altai mountains. Three Deniso-van mitochondrial genomes have been generated from material in the cave, dated by poorly associated fauna (5) at more than 50,000years old. The diversity of these genomes indicates that the Denisovan population hada larger long-term average size than that of the Neandertals (6 ,7), suggesting that the Denisovans were formerly widespread across mainland East Asia. However, interbreeding with modern humans only appears to have occurred in remote Island Southeast Asia,requiring marine crossings and raising questions about the distribution and fossil record of Denisovans in Island Southeast Asia.The distribution of modern human populations containing detectable amounts of introgressed Denisovan DNA is surprising, as none have been detected in main-land Asia (introgressed DNA refers to small amounts of DNA from one species found in another species). Denisovan DNA has only been found on islands east of Wallace’s Line(see the figure). The modern human populations with the highest percentage of Den-isovan DNA are the geographically isolated New Guinean and Australian aborigines (~3to 4%) (4), whereas smaller percentages have been detected in a range of populations in Island Southeast Asia. Groups in this area arethought to be descended from early Southeast Asian hunter-gatherers and later Neolithic farmers (3). Wallace’s Line (8) is one of the world’s biggest biogeographic disjunctions, mark-ing the border of placental-dominated ecosystems to the west, whereas the lesser knownLydekker’s Line marks marsupial-dominatedecosystems to the east (see the figure). Onlytwo terrestrial mammal groups are knownto have crossed Wallacea (the area betweenthe two lines) to migrate into Australasia:rodents and anatomically modern humans.The discovery of Homo floresiensis(“Hob- bits”) on Flores in 2003 (9) indicates a sepa-rate dispersal across Wallace’s Line, whereasa ~67,000-year-old foot bone from Callao in the Philippines represents a small-bodiedhominin of unknown taxonomic affiliation(10). These taxa remain enigmatic, but suggest that other hominin species had the capacity to cross the powerful marine current that forms and maintains Wallace’s Line even during times of lowered sea levels. Denisovan populations appear to have had a diverse ecological range covering both mainland and Island Southeast Asia (5). The inferred large historical population size is consistent with the use of the extensive savannah regions on the exposed Sunda shelf as are fugium during Pleistocene glacial phases (11). The exposed shelf would have allowed northward and southward migration during climatic cycles.The location of the Denisovan reference specimens in the Altai mountains might suggest that Denisovan DNA gene flow into mod-ern human populations occurred somewhere on the Asian mainland, before spreading throughout the Southeast Asian region. Theapparent absence of Denisovan introgression in current mainland populations is most easily explained through overwriting by the DNAof incoming East Asian populations in areas other than Island Southeast Asia. However,analysis of indigenous negrito/hunter-gath-erer populations on mainland Malaysia and the Andaman Islands revealed no DenisovanDNA introgression, even though the long-isolated Andaman Islanders show no admixture with other East Asian populations (3). Simi-larly, genomic analysis of an ancient modern human in China (Tianyuan, ~40,000 yearsold) detected no Denisovan DNA (12), argu-ing against the existence of a prehistoric inter- breeding signal that has been overwritten.Together, these observations argue against an ancient introgression of Denisovan DNAon the Asian mainland (3). Instead, the source of the Denisovan gene flow appears to have been east of Wallace’s Line, with the lack of Denisovan DNA in mainland populationsexplained by Wallace’s Line limiting the reverse dispersal of introgressed populations.Subsequent movements of East Asian/Neolithic modern humans appear to have diluted the Denisovan-introgressed populations out-side Australia and New Guinea, and also carried the signal further throughout the area andacross the Pacific (3). 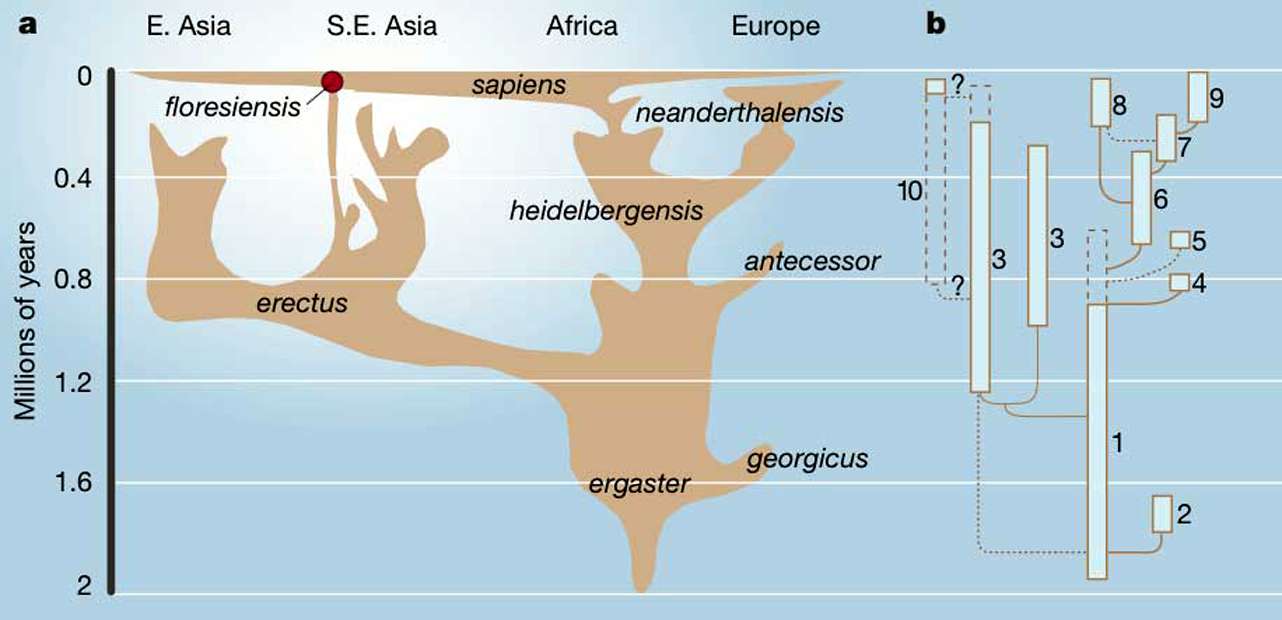 The only well-characterized hominin known to have crossed Wallace’s Line beforemodern humans is H. floresiensis, whose affinities remain enigmatic, although morphological analyses suggest derivation from an early Homo erectus ancestor or an even more primitive species (9). A stone toolrecord on Flores dated to more than 1 million years supports an early presence (9). How-ever, despite its location beyond Wallace’sLine, trying to identify H. floresiensis as aregional representative of the Denisovans isdifficult to reconcile with the enlarged molarsof the Denisovans, and the divergence date of Denisovans and modern human populations estimated with mitochondrial DNA at ~1million years ago (1), or 170,000 to 700,000years ago with genomic data (4).The considerable difference between these divergence date estimates is interest-ing, and may relate to recent reports that the Denisovan genome contains large amounts of introgressed Neandertal genomic DNA (6 ,7). This will affect estimates of both the phyogenetic relationships and genomic diver-gence dates between Neandertals, Deniso-vans, and modern humans. Alternatively, the older mitochondrial divergence date of ~1 million years may reflect the input of a more ancient Asian population, or that all the dates are overestimates resulting from the temporal dependency of molecular rates, and the erroneously low rate produced by the distant chimp-human external calibration (13). We thus infer that H. floresiensis was an endemic species whose lineage originated at least 1 million years ago, restricted to a small region of Wallacea, whereas the Denisovans probably arrived during the mid-Pleistocene (after 600,000 years ago) and spread more widely in the region. The Denisovans east of the Wallace line may be represented by the Philippines Callao specimen, or have not yet been recognized. Other enigmatic hominin remains in Asia—from Narmada (India)and Dali, Jinniushan, Maba, and Xujiayao(China)—may represent the apparently once more extensive Denisovan population, or per-haps yet other species The Denisovan genome reportedly alsocontains a small contribution from another archaic population, whose source is cur-rently unknown (6 ,7). Did the Denisovansinterbreed with a more ancient species, such as H. erectus or H. antecessor , or perhaps a late surviving H. heidelbergensis in Asia(14)? Given the uncertainties in the molec-ular dates, the genomic divergences may be compatible with a recent model suggestingthat modern humans, Neandertals, and Densovans are a trichotomy that originated from the widely dispersed Middle Pleistocenespecies H. heidelbergensis (14). The fragmentary and disparate nature of the East Asian fossil record provides only tantalizing glimpses of a diversity of hominin groups. Similarly, the apparently widespread distribution of early hominins across Wallacea, exemplified by the finds from Flores and the Philippines, raisesthe issue of whether they could even have extended to the Sahul shelf and regions like New Guinea and Australia (see the figure).Why did gene flow between Denisovans and modern human populations occur primarily east of Wallace’s Line and not on the Asian mainland? Given that intentional dispersal to Wallacea required the use of watercraft, the first modern human groups encountering the established Denisovan populations were likely to have been of very limited size. Either interbreeding may be more likely under these circumstances, or any interbreeding that does occur is more likely to be preserved as a signal in descen-dants. The genomic evidence suggests that gene flow from the Denisovans may have been largely male-mediated, providing someclues about the nature of the interactions(4). In addition, rapid dispersal by mod-ern humans into tropical Wallacea is like lyto have led to exposure to a wide range of new pathogens, such that disease resistance alleles obtained through hybridization with native populations may have been selec-tively advantageous (15). The first groups of modern humans leaving Africa, whichwere also presumably of limited size, sim-ilarly appear to have interbred during ini-tial encounters with established Neandertal populations in western Asia (16). An anticipated wealth of new genomic data are set to further illuminate the nature of these inter-actions between Neandertals, Denisovansand modern humans, as well as the extent and possible functionality of the DNA that was exchanged. References and Notes1. J. Krause et al., Nature 464, 894 (2010). 2. D. Reich et al., Nature 468, 1053 (2010). 3. D. Reich et al., Am. J. Hum. Genet. 89, 516 (2011). 4. M. Meyer et al., Science 338, 222 (2012). 5. A. Gibbons, Science 333, 1084 (2011). 6. S. Pääbo, The Biology of Genomes, Cold Spring Harbor, 7to 11 May 2013. 7. E. Pennisi, Science 340, 799 (2013). 8. T. H. Huxley, Proc. Zool. Soc. Lond. 1868, 296 (1868). 9. M. J. Morwood, W. L. Jungers, J. Hum. Evol. 57, 640(2009). 10. A. S. Mijares et al., J. Hum. Evol. 59, 123 (2010). 11. J. R. Stewart, C. B. Stringer, Science 335, 1317 (2012). 12. Q. Fu et al.,Proc. Natl. Acad. Sci. U.S.A. 110, 2223(2013). 13. S. Y. W. Ho et al., Mol. Ecol. 20, 3087 (2011). 14. C. Stringer, Evol. Anthropol. 21, 101 (2012). 15. L. Abi-Rached et al., Science 334, 89 (2011). 16. R. E. Green et al., Science 328, 710 (2010). 17. J. Balme, I. Davidson, J. McDonald, N. Stern, P. Veth, Quat. Int. 202, 59 (2009). 18. J. F. O’Connell, J. Allen, K. Hawkes, in The Global Originsand Development of Seafaring (McDonald InstituteMonographs, Cambridge, UK, 2010), pp. 57–68. Cooper, Alan, and C. B. Stringer. "Did the Denisovans Cross Wallace's Line?." Science 342.6156 (2013): 321-323. |
|
|
|
Post by Admin on Jul 22, 2014 22:31:17 GMT
The “Rhineland hypothesis” envisions modern European Jews to be the descendents of the Judeans—an assortment of Israelite–Canaanite tribes of Semitic origin (figs. 1 and 2) (supplementary note S1, Supplementary Material online). It proposes two mass migratory waves: the first occurred over the 200 years following the Muslim conquest of Palestine (638 CE) and consisted of devoted Judeans who left Muslim Palestine for Europe (Dinur 1961). Whether these migrants joined the existing Judaized Greco–Roman communities is unclear, as is the extent of their contribution to the Southern European gene pool. The second wave occurred at the beginning of the 15th century by a group of 50,000 German Jews who migrated eastward and ushered an apparent hyper-baby-boom era for half a millennium (Atzmon et al. 2010). The Rhineland hypothesis predicts a Middle Eastern ancestry to European Jews and high genetic similarity among European Jews (Ostrer 2001; Atzmon et al. 2010; Behar et al. 2010).  FIG. 1.— Map of Eurasia. A map of Khazaria and Judah is shown with the state of origin of the studied groups. Eurasian Jewish and non-Jewish populations used in all analyses are shown in square and round bullets, respectively (supplementary table S3, Supplementary Material online). The major migrations that formed Eastern European Jewry according to the Khazarian and Rhineland hypotheses are shown in yellow and brown, respectively. FIG. 1.— Map of Eurasia. A map of Khazaria and Judah is shown with the state of origin of the studied groups. Eurasian Jewish and non-Jewish populations used in all analyses are shown in square and round bullets, respectively (supplementary table S3, Supplementary Material online). The major migrations that formed Eastern European Jewry according to the Khazarian and Rhineland hypotheses are shown in yellow and brown, respectively.The competing “Khazarian hypothesis” considers Eastern European Jews to be the descendants of Khazars (supplementary note S1, Supplementary Material online). The Khazars were a confederation of Slavic, Scythian, Hunnic–Bulgar, Iranian, Alans, and Turkish tribes who formed in the central–northern Caucasus one of most powerful empires during the late Iron Age and converted to Judaism in the 8th century CE (figs. 1 and 2) (Polak 1951; Brook 2006; Sand 2009). The Khazarian, Armenian, and Georgian populations forged from this amalgamation of tribes (Polak 1951) were followed by relative isolation, differentiation, and genetic drift in situ (Balanovsky et al. 2011). Biblical and archeological records allude to active trade relationships between Proto-Judeans and Armenians in the late centuries BCE (Polak 1951; Finkelstein and Silberman 2002), that likely resulted in a small scale admixture between these populations and a Judean presence in the Caucasus. After their conversion to Judaism, the population structure of the Judeo–Khazars was further reshaped by multiple migrations of Jews from the Byzantine Empire and Caliphate to the Khazarian Empire (fig. 1). Following the collapse of their empire and the Black Death (1347–1348) the Judeo–Khazars fled westward (Baron 1993), settling in the rising Polish Kingdom and Hungary (Polak 1951) and eventually spreading to Central and Western Europe. The Khazarian hypothesis posits that European Jews are comprised of Caucasus, European, and Middle Eastern ancestries. Moreover, European Jewish communities are expected to be different from one another both in ancestry and genetic heterogeneity. The Khazarian hypothesis also offers two explanations for the genetic diversity in Caucasus groups first by the multiple migration waves to Khazaria during the 6th–10th centuries and second by the Judeo–Khazars who remained in the Caucasus.  FIG. 2.— An illustrated timeline for the relevant historical events. The horizontal dashed lines represent controversial historical events explained by the different hypotheses, whereas solid black lines represent undisputed historical events. FIG. 2.— An illustrated timeline for the relevant historical events. The horizontal dashed lines represent controversial historical events explained by the different hypotheses, whereas solid black lines represent undisputed historical events.PCA was next used to identify independent dimensions that capture most of the information in the data. PCA was applied using two frameworks: the “multipopulation” carried for all populations (fig. 3) and separately for Eurasian populations along with Pygmies and Han Chinese (supplementary fig. S2, Supplementary Material online) and our novel “dual-population” framework (supplementary fig. S3, Supplementary Material online). In all analyses, the studied samples aligned along the two well-established geographic axes of global genetic variation: PC1 (sub-Saharan Africa vs. the rest of the Old World) and PC2 (east vs. west Eurasia) (Li et al. 2008). Our results reveal geographically refined groupings, such as the nearly symmetrical continuous European rim extending from Western to Eastern Europeans, the parallel Caucasus rim, and the Near Eastern populations (supplementary fig. S1, Supplementary Material online) organized in Turk–Iranian and Druze clusters (fig. 3). Middle Eastern populations form a gradient along the diagonal line between Bedouins and Near Eastern populations that resembles their geographical distribution. The remaining Egyptians and the bulk of Saudis distribute separately from Middle Eastern populations.  FIG. 3.— Scatter plot of all populations along the first two principal components. For brevity, we show only the populations relevant to this study. The inset magnifies Eurasian and Middle Eastern individuals. Each letter code corresponds to one individual (supplementary table S3, Supplementary Material online). A polygon surrounding all of the individual samples belonging to a group designation highlights several population groups. FIG. 3.— Scatter plot of all populations along the first two principal components. For brevity, we show only the populations relevant to this study. The inset magnifies Eurasian and Middle Eastern individuals. Each letter code corresponds to one individual (supplementary table S3, Supplementary Material online). A polygon surrounding all of the individual samples belonging to a group designation highlights several population groups.European Jews are expected to cluster with native Middle Eastern or Caucasus populations according to the Rhineland or Khazarian hypotheses, respectively. The results of all PC analyses (fig. 3, supplementary figs. S2 and S3, Supplementary Material online) show that over 70% of European Jews and almost all Eastern European Jews cluster with Georgian, Armenian, and Azerbaijani Jews within the Caucasus rim (fig. 3 and supplementary fig. S3, Supplementary Material online). Approximately 15% of Central European Jews cluster with Druze and the rest cluster with Cypriots. All European Jews cluster distinctly from the Middle Eastern cluster. Strong evidence for the Khazarian hypothesis is the clustering of European Jews with the populations that reside on opposite ends of ancient Khazaria: Armenians, Georgians, and Azerbaijani Jews (fig. 1). Because Caucasus populations remained relatively isolated in the Caucasus region and because there are no records of Caucasus populations mass-migrating to Eastern and Central Europe prior to the fall of Khazaria (Balanovsky et al. 2011), these findings imply a shared origin for European Jews and Caucasus populations.  FIG. 4.— Biogeographical origin of European Jews. First two principal components were calculated for Pygmies, French Basques, Han Chinese (black), Armenians (blue), and Eastern or Central European Jews (red)—all of equal size. PCA was calculated separately for Eastern and Central European Jews and the results were merged. Using the first four populations as a training set, Eastern (squares) and Central (circles) European Jews were assigned to geographical locations by fitting independent linear models for latitude and longitude as predicted by PC1 and PC2. Each shape represents an individual. Major cities are marked in cyan. FIG. 4.— Biogeographical origin of European Jews. First two principal components were calculated for Pygmies, French Basques, Han Chinese (black), Armenians (blue), and Eastern or Central European Jews (red)—all of equal size. PCA was calculated separately for Eastern and Central European Jews and the results were merged. Using the first four populations as a training set, Eastern (squares) and Central (circles) European Jews were assigned to geographical locations by fitting independent linear models for latitude and longitude as predicted by PC1 and PC2. Each shape represents an individual. Major cities are marked in cyan.The geographical origins of European Jews varied for different reference populations (fig. 4 and supplementary fig. S5, Supplementary Material online), but all the results converged to Southern Khazaria along modern Turkey, Armenia, Georgia, and Azerbaijan. Eastern European Jews clustered tightly compared with Central European Jews in all analyses. The smallest deviations in the geographical coordinates were obtained with Armenians for both Eastern (38 ± 2.7° N, 39.9 ± 0.4° E) and Central (35 ± 5° N, 39.7 ± 1.1° E) European Jews (fig. 4). Similar results were obtained for Georgians (supplementary fig. S5, Supplementary Material online). Remarkably, the mean coordinates of Eastern European Jews are 560 km from Khazaria’s southern border (42.77° N, 42.56° E) near Samandar—the capital city of Khazaria from 720 to 750 CE (Polak 1951). The duration, direction, and rate of gene flow between populations determine the proportion of admixture and the total length of chromosomal segments that are identical by descent. Admixture calculations were carried out using a supervised learning approach in a structure-like analysis. This approach has many advantages over the unsupervised approach that not only traces ancestry to K abstract unmixed populations under the assumption that they evolved independently (Chakravarti 2009; Weiss and Long 2009) but also is problematic when applied to study Jewish ancestry, which can be dated only as far back as 3,000 years (fig. 2). Moreover, the results of the unsupervised approach vary based on the particular populations used for the analysis and the choice of K, rendering the results incomparable between studies. Admixture was calculated with a reference set of seven populations representing largely genetically distinct regions: Pygmies (South Africa), Palestinians (Middle East), Armenians (Caucasus), Turk–Iranians (Near East), French Basque (West Europe), Chuvash (East Europe), and Han Chinese (East Asia) (fig. 5). The ancestral components grouped all populations by their geographical regions with European Jews clustering with Caucasus populations. As expected, Eastern and Western European ancestries exhibit opposite gradients among European populations. The Near Eastern–Caucasus ancestries are dominant among Central (38%) and Eastern (32%) European Jews followed by Western European ancestry (30%). Among non-Caucasus populations, the Caucasus ancestry is the largest among European Jews (26%) and Cypriots (31%). These populations also exhibit the largest fraction of Middle Eastern ancestry among non–Middle Eastern populations. As both Caucasus and Middle Eastern ancestries are absent in Eastern European populations, our findings suggest that Eastern European Jews acquired these ancestries prior to their arrival to Eastern Europe. Although the Rhineland hypothesis explains the Middle Eastern ancestry by stating that Jews migrated from Palestine to Europe in the 7th century, it fails to explain the large Caucasus ancestry, which is nearly endemic to Caucasus populations.  FIG. 5.— Admixture analysis of European, Caucasus, Near Eastern, and Middle Eastern populations. The x axis represents individuals from populations sorted according to their ancestries and arrayed geographically roughly from North to South. Each individual is represented by a vertical stacked column (100%) of color-coded admixture proportions of the ancestral populations. FIG. 5.— Admixture analysis of European, Caucasus, Near Eastern, and Middle Eastern populations. The x axis represents individuals from populations sorted according to their ancestries and arrayed geographically roughly from North to South. Each individual is represented by a vertical stacked column (100%) of color-coded admixture proportions of the ancestral populations.
Although they cluster with Caucasus populations (fig. 5), Eastern and Central European Jews share a large fraction of Western European and Middle Eastern ancestries, both absent in Caucasus populations. According to the Khazarian hypothesis, the Western European ancestry was imported to Khazaria by Greco–Roman Jews, whereas the Middle Eastern ancestry alludes to the contribution of both early Israelite Proto-Judeans as well as Mesopotamian Jews (Polak 1951; Koestler 1976; Sand 2009). Central and Eastern European Jews differ mostly in their Middle Eastern (30% and 25%, respectively) and Eastern European ancestries (3% and 12%, respectively), probably due to late admixture. Druze exhibits a large Turk–Iranian ancestry (83%) in accordance with their Near Eastern origin (supplementary fig. S4, Supplementary Material online). Druze and Cypriot appear similar to European Jews in their Middle Eastern and Western European ancestries, though they differ largely in the proportion of Caucasus ancestry. These results can explain the genetic similarity between European Jews, Southern Europeans, and Druze reported in studies that excluded Caucasus populations (Price et al. 2008; Atzmon et al. 2010; Zoossmann-Diskin 2010). Overall, our results portray the European Jewish genome as a mosaic of Near Eastern-Caucasus, Western European, Middle Eastern, and Eastern European ancestries in decreasing proportions. Dissecting uniparental haplogroups allows us to delve further into European Jews’ migration routes. As the results do not specify whether the Southern Europe–Caucasus migration was ancient or recent nor indicate the migration’s direction, that is, from Southern Europe to the Caucasus or the opposite, there are four possible scenarios. Of these, the only historically supportable scenarios are ancient migrations from Southern Europe toward Khazaria (6th–13th centuries) and more recent migrations from the Caucasus to Central and Southern Europe (13th–15th centuries) (Polak 1951; Patai and Patai 1975; Straten 2003; Brook 2006; Sand 2009). A westward migration from the diminished Khazaria toward Central and Southern Europe would have exhibited a gradient from the Caucasus toward Europe for both matrilineal and patrilineal lines. Such a gradient was not observed. By contrast, Judaized Greco–Roman male-driven migration directly to Khazaria is consistent with historical demographic migrations and could have created the observed pattern. Moreover, we found little genetic similarity between European Jews and populations eastward to the Caspian Sea and southward to the Black Sea, delineating the geographical boundaries of Khazaria (table 1 and fig. 1). Elhaik, Eran. " The missing link of Jewish European ancestry: Contrasting the Rhineland and the Khazarian hypotheses." Genome biology and evolution 5.1 (2013): 61-74. |
|

