|
|
Post by Admin on Jul 30, 2014 22:56:26 GMT
 The Dutch are descended from a group of Homo sapiens who settled Europe during the Paleolithic (40 000 years ago) and Neolithic (10 000 years ago) periods.15 These settlers originated in the Middle Eastern and brought a discrete set of Y chromosomal and mitochondrial haplotypes, Indo-European languages, agriculture, and pottery.16, 17, 18 The settlers were hunters, who lived in communities of about 50 people at a population density of one person per 15–50 km2.15 This means that each community controlled an area of 750–2500 km2. Over the territory of the present Netherlands (41 536 km2), there must have been about 3000 inhabitants (Figure 1).  Figure 1. Population growth in the Netherlands: 10 000 BC to 2000 AD, indicating the major demographic events. Figure 1. Population growth in the Netherlands: 10 000 BC to 2000 AD, indicating the major demographic events.Agriculture supported higher population densities so that, in the early Bandkeramik period, there were about two inhabitants per square kilometer15 Over the territory of the present Netherlands, there must have been about 80 000 inhabitants. Several new cultures appeared around this time period of 5000–1000 BC, with the Megalithic monuments as the most prominent examples.19 Those tombs were made of large stones shaped as domens or menhirs, and appeared in the farming regions along the Atlantic coast of Europe from the Iberian Peninsula to Denmark, and along the western coasts of England and Ireland. Stonehenge in England is the most famous example. In the first millennium BC, the Netherlands were invaded by Celts, a people who were successful farmers and excellent copper smiths.20 They were descendants of the Iron Age communities that were named for Halstatt in western Austria and La Tène in Switzerland. During the next millennium, other groups settled in the Netherlands. Under Julius Caesar, the Romans invaded the Netherlands around the birth of Christ. A wave of Goths passed through the Netherlands around 400 AD, followed by Huns around 450 AD, and by the Germans around 800 AD (Figure 1). Regional sovereignty in the Netherlands began around 1000 AD. Immigration
In 1500, the population of the geographic region of the Netherlands counted between 900 000 and 1 000 000 inhabitants. This number increased to 1.85 million people in 1650, and, then, growth came almost to a standstill. In the 19th century, population growth resumed and, by 1880, the Dutch population increased to 2.1 million. By 1900, it reached 5.17 million, and by 1950 it doubled again to 10.2 million (Figure 1).21 International migration played a key role in shaping the Dutch population. From the late 16th century onward, large numbers of immigrants settled in the Netherlands, attracted by the religious tolerance and economic prosperity of the Dutch Republic.22, 23 After 1580, 100 000 religious and economic refugees fled from Flanders and northern France and settled with their families in the cities of the seaside provinces. A century later, 50 000 French Huguenots arrived in the Republic and, again, mostly settled in the cities and towns of the West. From the late 16th century until well into the 19th century, considerable economic migration occurred in the cities of the west, mainly from western Germany and Scandinavia. At the end of the 16th century, several thousand Jewish refugees from the Iberian peninsula arrived in the same area. From the second half of the 18th century onward, some 20 000 Jews arrived from Central and Eastern Europe. A rough estimate of the total 17th and 18th century migration into the Netherlands was on the order of half a million people, 60% of whom came from western Germany. Compared to a national population of two million people in 1800, these figures suggest that immigration was an important factor for shaping the demography of the Netherlands. Immigration declined after 1800 and did not become a demographic force again until the mid-20th century (Figure 1).22, 23 EmigrationEmigration was also a demographic force. Large numbers of indigenous Dutch people left the country during the past several centuries. In the 19th century, many Dutch people emigrated to other countries. During the first half of the 19th century, the number of Dutch émigrés living in the Dutch East Indies was around 2100 persons. Following the opening of the Suez Canal in 1869, that number increased to around 11 000 by the end of the 19th century. Emigration from the Netherlands to North America was not high during the 17th and 18th centuries – the number of people of Dutch descent in 1850 was only 10 000 – but after 1840, the flow increased. During the period 1830–1914, approximately 250 000 Dutch people emigrated to the United States. The first wave occurred between 1847 and 1857, with half of these persons belonging to a dissenting Protestant denomination. The second wave occurred later in the 19th century. The successive crop failures of potatoes and grain between 1847 and 1857, and the subsequent agrarian restructuring between 1880 and 1893, were influential push factors. Dutch migration to settlements in Brazil (1630–1654), Surinam (after 1667), and the Dutch Antilles was not extensive, and took place mostly in the 17th century, with a total of 15 000 emigrating up to 1900. Dutch emigration to settlements elsewhere was also very limited. Until 1920, about 11 000 Netherlanders settled in Canada, 4500 settled in Argentina, 3500 in Brazil, and 4000 in Australia. In 1914, 5000 Netherlanders lived in South Africa, a number that had been only 870 in 1891. After the Franco-Prussian War, when the German economy underwent a period of rapid expansion, many Dutch people went to Germany. The number of persons of Dutch descent living in Germany around 1860 was about 13 000, but by the outbreak of World War I, this had risen to more than 100 000. The number of persons of Dutch descent who had settled in Belgium in the mid-19th century was more than 30 000 and, after 1900, about 60 000. Regional and religious differencesDespite the relatively small size of the country, remarkable differences have always existed within the Netherlands, both by region and by religious group. The Netherlands has been divided geographically by the major rivers whose deltas they occupy – the Maas and the Rhine (Figure 2). Historically, these rivers created barriers to migration. Even greater geographic barriers were created for the people who lived on the West Frisian islands and for people who lived north of the Zuiderzee in Friesland (Figure 2, which describes the topographic situation around 1930 prior to the reclamation of the Zuiderzee).  Figure 2. Map of the Netherlands with mentioned provinces (uppercase only), cities and regions (upper and lowercase), and rivers (Maas and Rijn or Rhine). Figure 2. Map of the Netherlands with mentioned provinces (uppercase only), cities and regions (upper and lowercase), and rivers (Maas and Rijn or Rhine).Marital endogamy was determined not only by spatial isolation but also by religious differences. Sharp religious cleavages and vehement debates on religious problems have been characteristic of the Netherlands since the Reformation. In that crucial period, only a part of the population joined the Protestant faith. Both in the early 1830s, and in the 1880s, a number of ministers broke away from the Netherlands Reformed Church and returned to the stricter orthodoxy of an earlier period; these secessions led to the development of the 'Gereformeerde Kerken', the Calvinists. The percentage of Roman Catholics remained fairly constant (36.5% in 1869, 36.4% in 1930) and then increased slightly to 40.4% in 1971. The proportion of Dutch Reformed (the name that is given to the members of the original State church) declined continuously particularly after 1880, from 54.7 to 23.5% in 1971.27 The percentage of Calvinists (3% in 1869) rose to 9.4% in 1971. In its overall effect upon social and cultural participation, religion in the Netherlands was until recently of the utmost importance. From an early age onward, the population was divided into three separate blocs, who spent a great deal of their time in different environments. Religion determined the education that one received, the friends one had, and the choice of a spouse, the political party, and the trade union. Catholics and Calvinists, which constituted rather homogeneous and close-knit religious groupings with a marked identity of their own. The old distinctions between the southern provinces Limburg and North Brabant (85–95% Catholic), and the northern parts, with less than 10% Catholics, remained throughout the whole 19th and early 20th century. Outside the southern provinces, small concentrations of Roman Catholic groups could be found in a completely Protestant environment. This applied to parts of Friesland, Zeeuws-Vlaanderen in the province of Zeeland, Overijssel, Gelderland, and in the provinces of Holland (Figure 2). A detailed long-term study of religious assortative mating in the West of the Netherlands showed that religious barriers during the first decades of the 19th century were considerable by modern standards and became even more insurmountable after 1890.28 Provincial data for the period 1938–41 show that 89% of Roman Catholics married within their own religion. The Dutch Reformed and Calvinists had high endogamy rates in the Western provinces and Limburg, where both groups of Protestants were a small minority.29 National data for the period 1938–1983 showed a sharp increase in intermarriage for the Catholics and Calvinists, and a gradual increase among the Dutch Reformed.30 Religious endogamy in small communities was particularly high. Allele sharing with other European populationsThe Dutch share founder mutations with other European populations for many conditions, including acute intermittent porphyria,51 congenital disorders of glycosylation, type 1a,52 cystic fibrosis,53 familial hypercholesterolemia,43 generalized atrophic benign epidermolysis bullosa,54 glycogen storage disease II,55, 56, 57 Krabbe disease,58 medium chain acyl-coA dehydrogenase deficiency,59 Stargardt disease,60 and cone-rod dystrophy.60 (see Table 3) The frequency of these mutations as a cause of these diseases is variable, ranging from 4 to 94%. A single mutation is the major cause of the disease within the Netherlands, for generalized atrophic benign epidermolysis bullosa (COL17A1 2342delG),54 cystic fibrosis (CFTR deltaF508),49 Krabbe disease (GALC IVS10del30 kb),58 and MCAD deficiency (MCAD K304E).59 The Dutch/non-Dutch distinction may be arbitrary for many conditions, and may reflect that comparable studies for some of these mutations have not been performed in other European populations. Some of these mutations, including MCAD K304E,59 and CFTR deltaF508,53 show northwest to southeast clines across Europe. Dutch émigré populationsFounder mutations of Dutch origin have been found among the descendants of Dutch émigrés. Emigration was often by people who shared the same region of origin and the same religion, which could have had an effect on the genetic pool observed in the regions of destination. Unfortunately, no literature about founder mutations in the East Indies was identified. This may be caused by the fact that relatively little research on this topic has been performed in this country and that the Dutch were a nonstable, temporary group of migrants, who did not intend to settle there forever. During the independence of Indonesia, most Dutch descendents immigrated back to the Netherlands, limiting the possibility for Dutch genes to permeate the East Indies. The LDL receptor mutations of Dutch origin have been observed among Canadians with Dutch ancestry and among Afrikaners in South Africa (Table 4).43 Much like the Dutch themselves, Afrikaners are an endogamous population with Dutch, French Huguenot, and German ancestry. The multiple European origins have been observed for the Afrikaaner founder mutations for variegate porphyria PPOX R59W,66, 67 CFTR delF508, 394delTT, and 3272-26A right arrow G,62 the LDR receptor M408V,43 and the Fanconi anemia A deletion exons 12–31.63 Founders have been identified for mutations that cause Leber hereditary optic neuropathy (LHON) and acute intermittent porphyria in Dutch émigré populations. LHON is caused by mutation in the mitochondrial DNA (mtDNA). Recently, analysis of the 55 large Dutch pedigrees showed several instances in which the mtDNA mutations were either identical or related by descent.68 The most striking was a Dutch founder mtDNA 14484 mutation on a haplogroup J background that had an identical sequence in two Canadian LHON pedigrees, indicating a common ancestor. This mutation must have occurred prior to the European settlement of Canada in the 1600s. A total of 10 families with the 14484 mutation of the haplogroup J were traced to a female ancestor who was born in 1613 in the town of Rhenen (Figure 2). Another pedigree could be traced back to woman who was born in Rhenen in 1745 and another to a woman born around 1678 in Almkerk (Figure 2). In addition, there were two 14484/haplotype K pedigrees that could be traced to a woman born in Leeuwarden (Figure 2) circa 1760.68 Zeegers, Maurice PA, et al. " Founder mutations among the Dutch." European journal of human genetics 12.7 (2004): 591-600. |
|
|
|
Post by Admin on Aug 7, 2014 21:05:08 GMT
 Cut-marked bone (ulna) of Rock Dove specimens from Gorham’s Cave Credit: Ruth Blasco et al., Scientific Reports Feral Pigeons have colonised all corners of the Earth, having developed a close association with humans and their activities. The wild ancestor of the Feral Pigeon, the Rock Dove, is a species of rocky habitats, nesting typically on cliff ledges and at the entrance to large caves. This habit would have brought them into close contact with cave-dwelling humans, a relationship usually linked to the development of dwellings in the Neolithic. We show that the association between humans and Rock Doves is an ancient one with its roots in the Palaeolithic and predates the arrival of modern humans into Europe. At Gorham's Cave, Gibraltar, the Neanderthals exploited Rock Doves for food for a period of over 40 thousand years, the earliest evidence dating to at least 67 thousand years ago. We show that the exploitation was not casual or sporadic, having found repeated evidence of the practice in different, widely spaced, temporal contexts within the cave. Our results point to hitherto unappreciated capacities of the Neanderthals to exploit birds as food resources on a regular basis. More so, they were practising it long before the arrival of modern humans and had therefore invented it independently.  Figure 1 A) Location of Gorham's Cave, Gibraltar, in the southern Iberian Peninsula; (B) Top: General plan of Gorham's Cave showing the location of the excavated sectors [outer sector including the entrance and middle area of the cave, and inner sector (back of the cave)]; Bottom: Geological interpretative section of Gorham's Cave (NW-SE section or B-A projection in top) based on Jiménez-Espejo et al.7 and previous publications (e.g.9, 10, 12, 16); (C) Geological sequence of Gorham's Cave - left: schematic profile of the outer sector (middle area of the cave) modified from Collcut15 (see Barton et al.27 for more details); right: stratigraphic profile of the inner sector. Red boxes mark the archaeological levels/units studied here. The photograph in (A) was taken by C. Finlayson and the maps/graphs in (A, B) were made by J.R. and J.R.-V. by using CorelDRAW Graphics Suite 12 and CorelDRAW X3 software. We would like to acknowledge S.N. Collcut and R.N.E. Barton for the permission granted for the use of the geological sequence of Gorham's Cave shown in C-left, courtesy of the School of Archaeology, Oxford. Figure 1 A) Location of Gorham's Cave, Gibraltar, in the southern Iberian Peninsula; (B) Top: General plan of Gorham's Cave showing the location of the excavated sectors [outer sector including the entrance and middle area of the cave, and inner sector (back of the cave)]; Bottom: Geological interpretative section of Gorham's Cave (NW-SE section or B-A projection in top) based on Jiménez-Espejo et al.7 and previous publications (e.g.9, 10, 12, 16); (C) Geological sequence of Gorham's Cave - left: schematic profile of the outer sector (middle area of the cave) modified from Collcut15 (see Barton et al.27 for more details); right: stratigraphic profile of the inner sector. Red boxes mark the archaeological levels/units studied here. The photograph in (A) was taken by C. Finlayson and the maps/graphs in (A, B) were made by J.R. and J.R.-V. by using CorelDRAW Graphics Suite 12 and CorelDRAW X3 software. We would like to acknowledge S.N. Collcut and R.N.E. Barton for the permission granted for the use of the geological sequence of Gorham's Cave shown in C-left, courtesy of the School of Archaeology, Oxford.Gibraltar (36°7′N, 5°20′W) is located at the southern end of the Iberian Peninsula (Figure 1A). The western coast is sheltered by a wide bay, while the eastern side faces the Mediterranean Sea and is subjected to intense wave action. This particular phenomenon led to the creation and development of cavities of which Gorham's Cave is one7. Its sedimentary filling, from the deepest galleries to the present sea level, comprises a pure sandy aeolian record accumulated as a cliff-dune during climatic transgressive episodes. In the outermost area are frequent rock falls consisting of thin clast layers and seismic-like rock avalanches. In the inner sector the stratigraphy only displays local rock fall, aeolian dust, and karstic clay8. The first excavation was carried out at the outermost zone of the cave (entrance area) by Waechter9, 10 and was characterised by a low density of archaeological remains. The sediments were composed of interbedded and irregular-layered bright red clayey sand, reddish brown clay/sand with abundant charcoal flecks, dark yellowish brown compact clay/sand, and tank/pink sands and clays11, 12. Abundant limpets, other shells, and coarse, angular travertine roof falls occur within the lighter pink calcareous sandy units. The outer area was also excavated in its middle sector by Waechter9, 10 and later by Barton et al.13 and Stringer et al.14 (Figure 1B) and displays a lower sedimentation rate but also high human activity. These sediments tend to be somewhat less sandy than those in the outermost zone but are similar overall. In general, they include dark-brown organic-rich silty clay, grey sand, and irregularly bedded yellowish-brown sand containing coarse charcoal fragments; brown-black organic-rich clay with whitish gritty phosphatic lenses; and interbedded, massive, homogeneous, coarse brown sand (see Collcut15 for more detail). The inner sector was excavated at the beginning of this century by the Gibraltar Museum, and the first results were published by Finlayson et al.16. The excavations exposed an area of about 29 m2 of cave floor and produced a stratigraphy with 4 main occupation levels (I–IV from top to bottom). The sedimentary record in this zone is thinner than in the outer part of the cave due to the higher position of the bedrock. Likewise, its sedimentary composition differs from other sectors due to a predominance of clay minerals, calcite, and quartz, with small quantities of dolomite, ankerite, and feldspars7. The inner sedimentary series seems to register a more condensed record than the outer zone, making the stratigraphic and chronological correlation between excavation areas difficult. In the inner area, the chronology is based on a stratigraphically coherent series of AMS (Accelerator mass spectrometry) radiocarbon dates obtained from charcoal fragments. Level III is dated between ca. 12,640 and 10,880 BP for the Magdalenian horizon (level IIIa) and between ca. 18,440 and 16,420 BP for the Solutrean horizon (level IIIb). Level IV is dated between ca. 32,560 and 23,780 BP15. In the outer area, a combination of AMS radiocarbon and OSL dates is used to locate chronologically the outer deposits. Radiocarbon results reported by Higham et al.17 indicate an age of between ca. 29 and 51 kyr BP for UBSm.7 and BeSm.1. Nevertheless, the dates from the underlying LBSmff.1–5 of between ca. 42 and 56 Kyr BP suggest that most of the charcoal fragments from UBSm.7 and BeSm(OSsm) could have been moved up by the soft sediment loading17. The Single Grain (SG) OSL chronology and the Bayesian age model show sediments of MIS 5 age near the base of the sequence (119,300 ± 14800 kyr for CSm), with deposition occurring into early to mid MIS3 (48,700 ± 4000 for BeSm [PLSsm].3 and 38,500 ± 5800 kyr for UBSm.6)18. The lowest layer studied here (SSLm [Usm].5) yielded an OSL age of 67,900 ± 5150 kyr18.  In the middle and lower layers of the outer stratigraphic sequence, lithic tools are consistent with the Middle Palaeolithic techno-complexes. The knapping technique follows discoid reduction sequences, although a significant increase of laminar flakes coming from bipolar Levallois cores can be detected at SSLm.5–6 (MIS 4). Lithic tools belonging to the Upper Palaeolithic were identified from CHm.5 (unite D of Waechter10; see Collcut and Currant19 for correlations). In the inner sector, level IV corresponds to a Mousterian horizon characterized by the use of flint and fine-grained sandstone and to a lesser extent quartzite, quartz, and dolomite. These materials are usually exploited following Levallois and discoid reduction sequences. In some cases, the obtained flakes are configured as scrapers, side-scrapers, and denticulates. A significant change can be detected at level III, which is characterized by a blade technology, plain retouch, and the configuration of artefacts that could be classified into the Upper Palaeolithic techno-complexes with diagnostic pieces attributable to the Solutrean and Magdalenian20, 21. The faunal record in Gorham's Cave is typically European (i.e., without African influences) and fairly constant in taxonomical composition through the sequence with no marked fluctuations of species per archaeological units, especially in the case of macro-mammals22. The majority of identified ungulate remains belong to two species – Cervus elaphus and Capra ibex. This apparent stability of the environment surrounding the cave supports the hypothesis that southern Iberia did not suffer the extreme cold of glaciations or the aridity potentially generated by it. Only the occurrence of Grey Seal (Halichoerus grypus) registered by Sutcliffe in the D unit (CHm in Currant system; see Collcut and Currant19 for correlations) can be interpreted as punctual evidence of a cold phase since this species has never been previously recorded so far south22; this presence may simply reflect conditions to the north that may have forced some marine species south and not actual conditions on site. The amphibian and reptile assemblages from the inner part of the cave involve at least 24 taxa, including newts, toads, frogs, tortoises, turtles, lacertid and scincid lizards, geckos, and several snakes. These findings show an increase in the atmospheric temperature range during the latest Pleistocene, mainly due to lower winter temperatures23. The largest assemblage in the outer area comes from LBSmcf.11, which yielded 21 species with the southern spadefoot toad (Pelobates cultripes) as the most frequent taxa24. The small mammal record is remarkably stable with five dominant species – Oryctolagus cuniculus, Apodemus sylvaticus, Eliomys quercinus, Microtus brecciencis and Pitymys (Microtus) duodecimcostatus. In addition to these taxa, a selection of other species occurs in lower proportions though widely distributed through the stratigraphic sequence25.  Figure 2: Cut-marked bones of Rock Dove specimens from Gorham's Cave: sternum (A), ulna (B, E) and humerus (C, D) from level IV, and tibiotarsus from LBSmcf.2 (F). Figure 2: Cut-marked bones of Rock Dove specimens from Gorham's Cave: sternum (A), ulna (B, E) and humerus (C, D) from level IV, and tibiotarsus from LBSmcf.2 (F).Here, we examined 1,724 Rock Dove bones from inner [III and IV levels] and outer [BeSm (Ossm).1 to SSLm (Usm).5] areas of Gorham's Cave (Figure 1C; Table S1; Table S2), spanning the time range from 67 kyr to 28 kyr16, 27. This temporal range coincided with the occupation of the cave by Neanderthals and, subsequently, by modern humans. Twenty discrete archaeological units were examined taphonomically. Nineteen of these contexts were associated with Neanderthals and one with modern humans (level III from inner area)16, 27. We found evidence of human intervention on Rock Dove bones in 11 (57.89%) of the Neanderthal contexts, as well as in the modern human context (Table 1). There was no observable difference in the tendency of damaged bones between Neanderthals and modern humans, both of whom appeared to have regularly processed Rock Doves, presumably for food. In the case of the Neanderthal occupation units, we detected cut-marks on 28 dove bones, 16 from level IV and 12 from the outer area (Table 1; Table S3; Figure 2). Incisions tend to occur both on the wing (n = 22 of 992 or 2.22%) and lower limb bones (n = 5 of 282 or 1.77%), as well as on one sternum fragment (16.67%) (Table S3; Figure 2). Although the proportion of cut-marked specimens is not high, it is important to emphasize that the size of these prey makes the use of stone tools unnecessary for direct consumption. After skinning or feather removal, direct use of hands and teeth would be the best way to remove the meat and fat/cartilage from the bones28, 29. The proof of this is the human tooth-marks and associated damage observed on some dove bones (n = 15 of 1364 or 1.1%). These imprints and alterations resulting from disarticulation and/or direct consumption (bone breakage by overextension, e.g., arrachement and peeling) match up with the anthropogenic alterations described both experimental and archaeologically by Lefèvre and Pasquet30, Higgins31, and Laroulandie32, 33, 34. In addition, a proportion of the bird specimens show signs of burning (n = 158 of 1364 or 11.58%), some of them with double colouring evidence (n = 29 of 158 burnt bones or 18.35%) (Table S4). The latter alterations are due to the fact that the entire surface of the bone would not have been exposed to fire with the same intensity. This happens when the prey or portions of it (skinned or not) are placed on a fire place for roasting. The areas of the bone that have not meat on them (or only a very thin tissue), are affected by the heat more intensely, and therefore the degree of burning on these areas is higher. In contrast, the bone areas covered with large muscle mass remain unmodified or are modified only slightly, acquiring lower degrees of colouring. At Gorham's Cave, the double colouration on the dove bones coincides with the areas of the skeleton with low muscle mass. Thus, the highest grade of burning of the humerus is detected on the head of the proximal joint, on the distal end of the tibiotarsus and ulna, and on the distal part of the palmar surface of the radius. This type of evidence was also documented in the early Middle Palaeolithic of Bolomor Cave (Spain)28, 35, 36 and in sites with more recent chronologies, such as in the Upper Magdalenian site of La Vache (France)37 and at the Final Epigravettian levels of Grotta Romanelli (Italy)38. In these sites the presence of double colorations was interpreted as the result of birds being cooked over a fire or in burning embers. Although this practice seems to have been used by the Neanderthals of Gorham's Cave, the highest proportions of completely burnt bones belong to degree 2 (brown colour) and 3 (black colour) (n = 139 of 158 or 87.97%). It must be taken into account that burning on bone fragments might reflect other types of intentional activities, such as the removal of waste for cleaning purposes, or could be the result of unintended processes, such as accidental burning. It could even be a consequence of post-depositional damage (e.g., secondary burning when fire places were set up on bones buried close to the surface). In the case of Gorham's Cave, it is possible that part of the burning might be the result of non-nutritive events that occurred after consumption and/or deposition. In contrast, modifications by other agents, such as carnivores, were negligible. Only 0.81% (n = 11) of all the elements showed marks by carnivore gnawing, and 0.22% (n = 3) showed damage due to digestive action by birds of prey. Only in layers LBSmcf.11 and LBSmcf.13 was there no evidence of human exploitation while carnivore modifications were documented. Carnivore damage was represented by a unique tooth-marked dove specimen in each layer. Despite this lack of anthropogenic damage on Columba bones, other ungulate taxa showed human processing in LBSmcf.11, which yielded one burnt cervid metatarsal fragment and a Cervus elaphus radius shaft fragment retoucher22. Besides, both LBSmcf.11 and LBSmcf.13 displayed a significant collection of lithic industry following discoid and Levallois reduction sequences39. Discussion and ConclusionsOur results demonstrate unequivocally that Neanderthals, and later on modern humans, consumed Rock Doves. Furthermore, this was not an isolated or casual behaviour as it affected a significant number of individual doves over a long temporal period. It points to the origin of an association, within the context of caves, which has persisted to the present day. Until now, the systematic exploitation of birds has been considered to be an exclusive and defining feature of modern human behaviour40, although recent evidence has pointed to the regular exploitation of raptors and corvids (for feathers) by Neanderthals in Gorham's Cave41. There is also evidence of the use of feathers in Grotta di Fumane (MIS 3, Italy)42 and of raptor claws in Combe-Grenal (MIS 5b, France) and Les Fieux (MIS 3, France)43. The exploitation of pigeons could date as far back as the Middle Pleistocene from evidence of level IV of Bolomor Cave with 2 butchered individuals of Columba sp. (MIS 5e, Spain)35, 36 and from UA25 of Lazaret Cave with one processed specimen of Columba livia (MIS 6, France)44, 45. These are, nevertheless, isolated events in comparison with the diachronic consistency observed at Gorham's Cave. The regular use of Rock Doves for food is presented here for the first time from 11 distinct Neanderthal occupation units from Gorham's Cave. The lack of anthropogenic damage on dove bones in the 8 remaining layers might be explained by diverse factors. A potential problem is that human activities on small prey do not always leave tell-tale physical evidence on all bones46. Apart from the fact that the lithic tool does not always come into contact with the bone, birds can be butchered (after skinning and de-feathering) using only the teeth and hands, which often makes it difficult to distinguish these alterations from those generated by other predators. In this connection, it must also be considered that differences in occupational patterns (mobility and site functionality), socio-cultural factors, and/or human behavioural diversity could condition the range of exploited species. That is, Neanderthals could have developed different subsistence strategies in the same landscape depending on behavioural variables, which are difficult to control archaeologically36. Taking all the variables with the potential to obscure human intervention signals, in the case of Gorham's Cave we interpret the 11 Mousterian occupation units with human use of pigeons as the strongest currently available evidence of a recurrent and systematic behaviour by Neanderthals in a specific area. However, ascertaining archaeologically the way Rock Doves were procured is difficult, as different methods could have left no trace in the fossil record. But even if birds may be perceived as elusive prey due to their flight capabilities, no technology needs to be invoked for their capture. Birds are forced to incubate their eggs in a fixed position, the nest, where the nestlings grow until they reach full adult size (for altricial birds such as the pigeon). This makes eggs, nestlings and brooding adults relatively easy to catch by hand by a moderately skillful and silent climber. Even roosting birds at night are practically defenseless against stealth predators because birds rely on vision and/or hearing for protection against predators, and contrary to mammals they cannot exploit olfactory cues. Ethnographic examples indicate that hunter-gatherers often catch wild birds as bushmeat47. To catch birds, modern humans in traditional societies use a variety of bird traps (typically snares or netting systems baited with food), sometimes in combination with the imitation of bird calls by direct vocalization or by using whistles to attract individuals from far away48, 49. In any case, active collection by hand or by different hunting techniques would not be mutually exclusive with the occasional scavenging of dead animals in the cliff. Nevertheless, their relatively high decomposition rate, i.e., rapid decomposition50, and the low incidence of carnivores detected from their bones could relate to immediate access by human groups to these animals. Even with this, it is possible that pigeons were easier to catch than other quick-flying animals. However, evidence of exploitation of avian species presumably harder to catch (e.g., raptors and corvids) was previously reported in Finlayson et al.41 for the same site. Similar observations were also made in other Neanderthal contexts of France and Italy42, 43. Some cut-marked species coming from these studies are, for example, Aquila chrysaetos, Gypaetus barbatus, Aegypius monachus, Gyps fulvus, Falco vespertinus, Milvus milvus, and Pyrrhocorax pyrrhocorax/graculus. This core of evidence provides clear proof that Neanderthal cognitive capacities to obtain different avian taxa were comparable to those of modern humans. Thus, the exploitation of pigeons reported here is further evidence that puts the Neanderthals' abilities on par with those of modern humans. The Rock Dove is a colonial and fast-reproducing species, making it an ideal candidate for sustainable exploitation. The nesting habit of the Rock Dove and its distribution in the landscape are practically unique in the Palearctic51: the species is highly colonial any time of the year in any kind of cliff or rocky outcrop, inland or on the coast6. Few bird species are comparable in numbers and ubiquity. Seabirds are colonial species which are invariably linked to coastal areas, and other colonial bird species, such as herons, ibises, flamingos, and waterfowl, breed and forage necessarily in wetlands; corvids are forest birds, and a few colonial species, such as Jackdaws or choughs, that may breed in rocky outcrops or cliffs do not seem capable of reaching such high population densities as doves52. Other colonial birds, such as starlings, sparrows, swifts, and swallows, are again highly seasonal in their breeding and too small in size to have become staple prey. Furthermore, pigeon nesting colonies and foraging flocks have practically no upper limit in size. Even today, millions of birds may flock together, as in Argentina53, and the largest flocks ever registered for any bird species were those of the now extinct passenger pigeon54. The original distribution range of the Rock Dove includes the mid-latitude belt in Eurasia, and the overlap with the Neanderthal range would have been extensive. Few species of birds had the potential to become so abundant within the distribution range of the Neanderthals. In short, it seems that the Neanderthals living in Gorham's Cave regularly took this bird for food, and it was one of a small suite of species with similar characteristics that would have guaranteed a stable food supply in the rocky environment of the Gibraltar landscape, as probably in many other parts of the Neanderthal geographical range. Traditionally in human history, the pigeon has been considered a symbol of peace, love, and fertility55, three attributes that are deeply interwoven. Its origins may well have been with the Neanderthals who exploited this very fertility in a way that allowed them to target them for food without depleting their numbers. More information: Paper: dx.doi.org/10.1038/srep05971 |
|
|
|
Post by Admin on Aug 11, 2014 22:05:08 GMT
The gene ASPM (abnormal spindle-like microcephaly associated) is a specific regulator of brain size, and its evolution in the lineage leading to Homo sapiens was driven by strong positive selection. Here, we show that one genetic variant of ASPM in humans arose merely about 5800 years ago and has since swept to high frequency under strong positive selection. These findings, especially the remarkably young age of the positively selected variant, suggest that the human brain is still undergoing rapid adaptive evolution.  Fig. 1. Worldwide frequencies of ASPM haplogroup D chromosomes (defined as having the derived G allele at the A44871G diagnostic polymorphism), based on a panel of 1186 individuals. Fig. 1. Worldwide frequencies of ASPM haplogroup D chromosomes (defined as having the derived G allele at the A44871G diagnostic polymorphism), based on a panel of 1186 individuals.Human ASPM has 28 exons with a 10,434–base pair open reading frame (1) (fig. S1).We resequenced the entire 62.1-kb genomic region of ASPM in samples from 90 ethnically diverse individuals obtained through the Coriell Institute and from a common chimpanzee (7). This revealed 166 polymorphic sites (table S1). Using established methodology (7), we identified 106 haplotypes. One haplotype, numbered 63, had an unusually high frequency of 21%, whereas the other haplotypes ranged from 0.56% to 3.3% (fig. S2). Moreover, this haplotype differed consistently from the others at multiple polymorphic sites (save for a few rare haplotypes that are minor mutational or recombinational variants of haplotype 63, as discussed later) (table S2). Two of these polymorphic sites are nonsynonymous, both in exon 18, and are denoted A44871G and C45126A (numbers indicate genomic positions from the start codon, and letters at the beginning and end indicate ancestral and derived alleles, respectively). These two sites reside in a region of the open reading frame that was shown previously to have experienced particularly strong positive selection in the lineage leading to humans (4) (fig. S1). The unusually high frequency of haplotype 63 is strongly suggestive of positive selection (8). We tested the statistical significance of the possibility of positive selection using coalescent modeling (7). The frequency of haplotype 63 is notably higher in Europeans and Middle Easterners (including Iberians, Basques, Russians, North Africans, Middle Easterners, and South Asians), as compared with other populations (table S1). We therefore focused on this group to take advantage of its relatively simple and homogeneous demographic structure (9). Because 7 of the 50 Europeans and Middle Easterners were homozygous for haplotypes 63, we tested the probability of obtaining 7 or more homozygotes (among 50)for a single haplotype across a 62.1-kb region containing 122 segregating sites (the number of polymorphic sites found in Europeans and Middle Easterners). The recombination rate and the gene conversion rate of the locus used in the test were obtained from our polymorphism data (7). For demographic history, we assumed a severe bottleneck followed by exponential growth (7) that is likely to be much more stringent than the bottleneck associated with the colonization of Eurasia (10). These parameters produced a highly significant departure from the neutral expectation (P G 0.00001). The simulation was repeated with a wide range of demographic histories (7), all of which produced highly significant results. We repeated the above tests on the entire Coriell panel, which also demonstrated strong significance. Finally, we repeated these tests using the inferred frequency of haplotype 63 (instead of the number of individuals who are homozygous for this haplotype) and again obtained significant statistical values. These data indicate that haplotype 63 has spread to high frequency under positive selection. The two nonsynonymous polymorphisms previously mentioned are either the target of selection or closely linked to the target site. We define haplogroup D (where D stands for Bderived[) as the class of haplotypes with the derived G allele at the A44871G nonsynonymous polymorphic site. The haplotypes with the A allele are defined as non-D. This classification is meant to capture the notable structure in the 106 haplotypes. Haplogroup D comprises two subgroups. One contains the predominant haplotype 63 and its closely related mutational variants Eincluding haplotypes 64, 66, and 71 (table S2)^. The other contains recombinant haplotypes between haplotype 63 (or its close mutational variants) and non-D haplotypes (including haplotypes 62, 65, and 105). The frequency of haplogroup D chromosomes is 28% in the entire Coriell panel and 44% in Europeans and Middle Easterners. Another prominent feature separating D and non-D haplotypes is the fact that the two classes have fixed differences relative to each other at multiple sites, where D haplotypes have the derived alleles (except for the rare recombinants between D and non-D chromosomes). This unusual haplotype structure is consistent with the following evolutionary history: (i) a rapid increase of haplotype 63 from a single ancestral copy to high frequency, and (ii) the introduction of minor variants of haplotype 63 by mutation and by recombination between D and non-D chromosomes (these variants make up the other members of haplogroup D). Another signature of positive selection is extended linkage disequilibrium (LD) (8), which is evident in the 62.1-kb region. Indeed, the 50 haplogroup D chromosomes show nearly complete LD across the region, with only three cases of LD breakdown Ehaplotypes 62, 65, and 105, present in one, two, and one copy, respectively (table S2)^. In contrast, the non-D chromosomes do not show any unusual LD across the region. We investigated the decay of LD beyond the 62.1-kb region by sequencing the Coriell panel for two flanking intergenic segments of roughly 5 kb each, positioned 25 kb upstream and downstream of ASPM. There is notable LD breakdown in these two segments, confirming that the site of selection is most likely within ASPM. Following established methodology (7), we estimated the coalescence age (i.e., time to the most recent common ancestor) of haplogroup D at 5800 years, with a 95% confidence interval between 500 and 14,100 years. In comparison, the coalescence age of all the chromosomes (both D and non-D) is ~800,000 years. Thus, the age of haplogroup D substantially postdates the emergence of anatomically modern humans, estimated at ~200,000 years ago (11). A rough calculation showed that the fitness advantage of haplogroup D is in the range of a few percent over non-D haplotypes. By genotyping the A44871G diagnostic polymorphism described earlier, we obtained the global frequency distribution of haplogroup D chromosomes from a separate panel of 1186 individuals (7). Consistent with the Coriell panel, we observed much higher frequency of haplogroup D chromosomes in Europeans and Middle Easterners than in other populations (Fig. 1). The corresponding estimate of FST, a statistic of genetic differentiation, is 0.29 between Europeans/Middle Easterners and other populations and 0.31 between Europeans/Middle Easterners and sub-Saharan Africans. These values indicate considerable genetic differentiation at this locus (12). Several scenarios may account for such notable differentiation. One is that haplogroup D first arose somewhere in Eurasia and is still in the process of spreading to other regions. The other is that it arose in sub-Saharan Africa, but reached higher frequency outside of Africa partly because of the bottleneck during human migration out of Africa. Finally, it is possible that differential selective pressure in different geographic regions is partly responsible. Collectively, our data offer strong evidence that haplogroup D emerged very recently and subsequently rose to high frequency under strong positive selection. The recent selective history of ASPM in humans thus continues East (14)—the significance of this correlation is not yet clear. Mekel-Bobrov, Nitzan, et al. " Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens." Science 309.5741 (2005): 1720-1722. |
|
|
|
Post by Admin on Sept 3, 2014 21:25:14 GMT
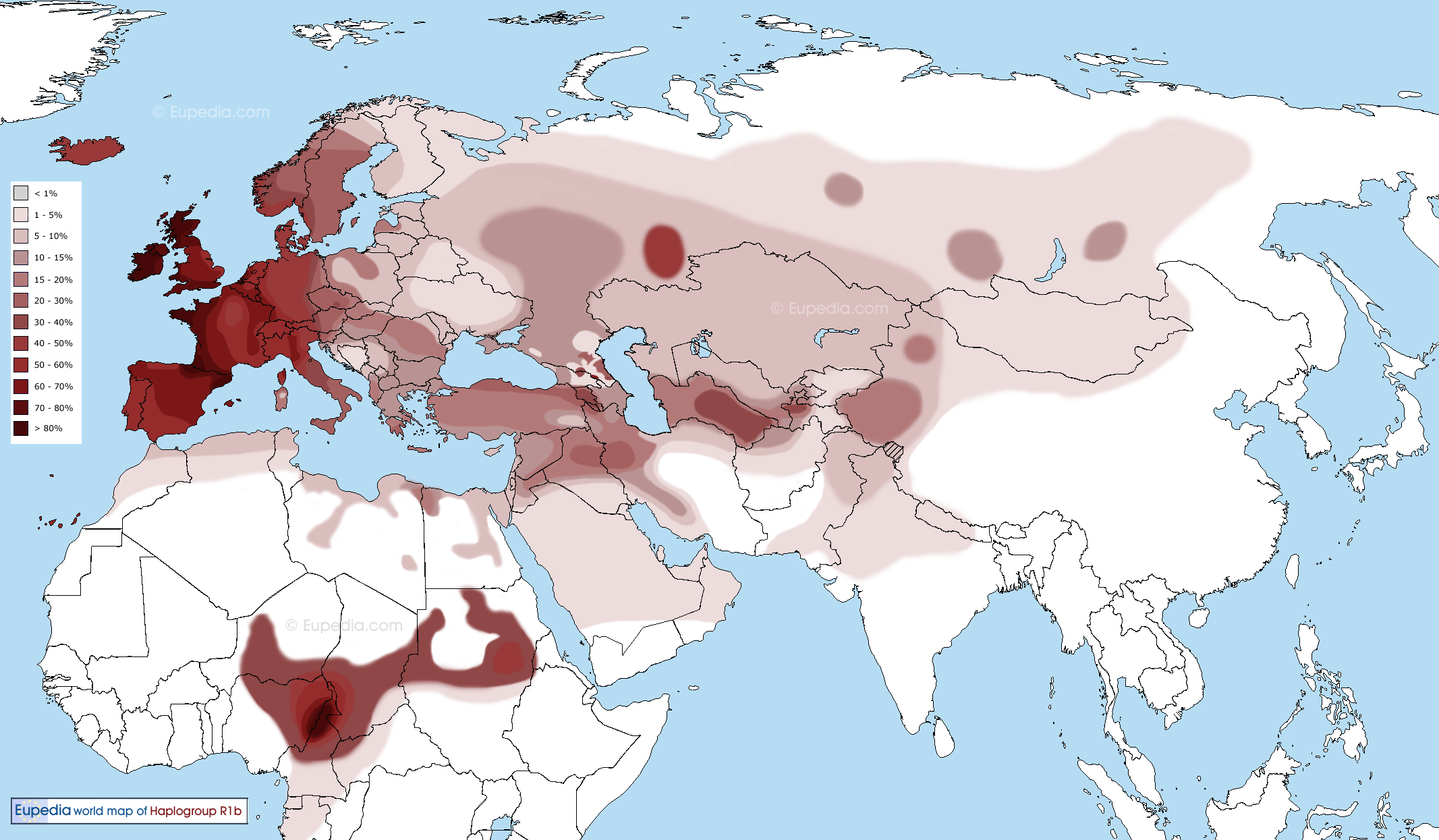 Human Y chromosomes belonging to the haplogroup R1b1-P25, although very common in Europe, are usually rare in Africa. However, recently published studies have reported high frequencies of this haplogroup in the central-western region of the African continent and proposed that this represents a ‘back-to-Africa’ migration during prehistoric times. To obtain a deeper insight into the history of these lineages, we characterised the paternal genetic background of a population in Equatorial Guinea, a Central-West African country located near the region in which the highest frequencies of the R1b1 haplogroup in Africa have been found to date. In our sample, the large majority (78.6%) of the sequences belong to subclades in haplogroup E, which are the most frequent in Bantu groups. However, the frequency of the R1b1 haplogroup in our sample (17.0%) was higher than that previously observed for the majority of the African continent. Of these R1b1 samples, nine are defined by the V88 marker, which was recently discovered in Africa. As high microsatellite variance was found inside this haplogroup in Central-West Africa and a decrease in this variance was observed towards Northeast Africa, our findings do not support the previously hypothesised movement of Chadic-speaking people from the North across the Sahara as the explanation for these R1b1 lineages in Central-West Africa. The present findings are also compatible with an origin of the V88-derived allele in the Central-West Africa, and its presence in North Africa may be better explained as the result of a migration from the south during the mid-Holocene. Characterisation of the male lineages of Equatorial GuineaIn the 112 samples analysed, we were able to identify 104 different haplotypes and 13 different haplogroups (Figure 1 and Supplementary Table S2). The majority of the Y-SNP lineages found in this study (almost 80%) belong to haplogroup E, namely bearing the M2-derived allele, the most common haplogroup in sub-Saharan Africa Bantu populations. Apart from this haplogroup and five other chromosomes, all the remaining samples belong to haplogroup R, namely R1b1-P25, a lineage that is rare in Africa and is found mainly in Europe and Asia. Nevertheless, high frequencies of these lineages in some African populations have been previously reported by several authors.5, 6, 7, 29, 30 Lineages in clade A, although almost entirely restricted to Africa, have been described in Bantu populations at low frequencies. These lineages are mostly present in Nilo-Saharan speakers (for example, reference2, 5, 6, 21) and therefore, a high frequency was not expected in our sample. This expectation was confirmed; only one of the chromosomes in our sample belongs to this haplogroup, more specifically to the haplogroup A3b2-M13, which is more frequently observed among Nilotes than other African groups.3 Similarly, only one chromosome in our sample was found to belong to haplogroup B, namely to the branch that is most common in Bantu individuals (defined by the derived allele at M150).2, 5, 18, 21  The major haplogroup E, the most diverse clade in the Y chromosome tree, is widespread across the African continent, where its highest frequencies are found and is also present in the Middle East, southern Europe, and Central and South Asia.17 This clade was the most frequent in our sample, representing almost 80% of all the chromosomes, similar to results for other sub-Saharan populations (for example, reference5, 30). The most frequent sub-lineage found in our sample inside this clade was the haplogroup E1b1a-M2, proposed as a marker of Bantu expansion.22, 30 Nevertheless, some other typically non-Bantu lineages were also found in our sample. One is the E1a-M33 haplogroup, which is usually not found further South in the Bantu expansion route (for example, Angola18) but is detected at higher frequencies above the starting point of these migrations (for example, Guinea-Bissau31). Therefore, the presence of this lineage could be an indication of a genetic pool that existed before the beginning of the Bantu expansion and was not spread by this movement. Other non-Bantu lineages found within this haplogroup are E1b1b1a-M78 and E1b1b1-M81, which have been frequently found in North Africa and also in Europe.32 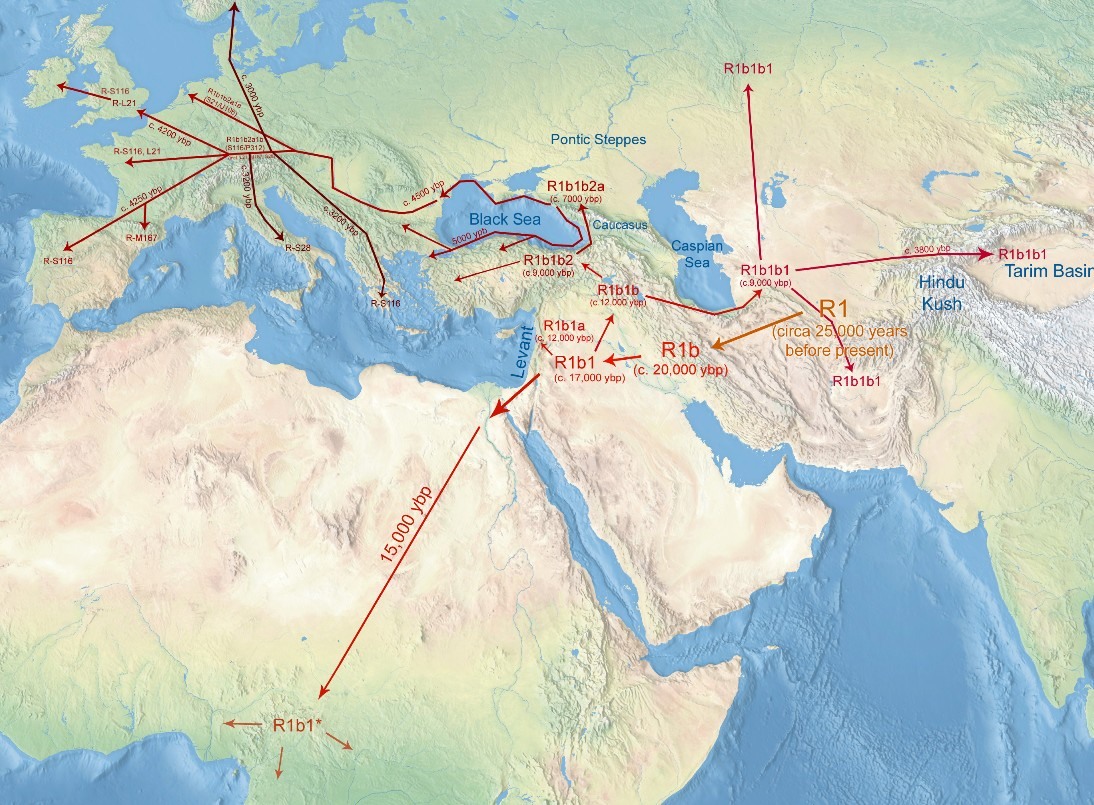 Both haplogroups G and N (accounting for two and one chromosomes, respectively) are rare in Africa, and their low frequency in this sample can most likely be explained by a recent Eurasian influx. Altogether, the proportion of recent Eurasian admixture found in our sample is approximately 15% (haplogroups E1b1b1b-M81, G-M201, N1c-Tat, R1b1b2-M269 and two chromosomes belonging to E1b1b1a-M78—see criteria below), which is easily explained by the well-reported European arrivals to this territory within the last five centuries. For the Y-SNP data, the haplogroup diversity of our sample (0.7526±0.0261), although higher than that found in other West African populations,18 is still lower than for other regions, a fact that has been explained by a loss of diversity during the Bantu migrations.3, 5, 18 For the Y-STRs, high levels of haplotype diversity (0.9987±0.0014) and of the mean number of pairwise differences (9.5048±4.3932) were found. The results obtained from Equatorial Guinea were compared with available data for other African populations5, 33, 34, 35 (see Supplementary Figure S2 and Table 1). However, Africa is still a poorly studied continent, and data for comparative analysis were not always accessible. Furthermore, the data for all populations need to be reduced to the same resolution level, requiring a minimal common set of Y-STRs and further constraining the amount of data available for comparisons. To overcome potential bias due to recent historical factors (for example, colonisation by Europeans), the levels of genetic diversity were calculated in different African samples and within each lineage (with n>5) after excluding chromosomes that had most likely been recently introduced by Europeans (including haplogroups E1b1b1, F, G, N1c, R1a, R1b1b2 and T). Because chromosomes belonging to haplogroup E1b1b1a-M78 can be considered of either African or European ancestry, a search for identical haplotypes was performed on the YHRD,36 and only those that did not present a match in European populations were maintained in the analysis.  Figure 2.. Phylogenetic network constructed with information from (a) 10 Y-STR haplotypes (DYS19, DYS389I, DYS389II, DYS390, DYS391, DYS392, DYS393, DYS437, DYS438 and DYS439) within the R1b1-P25 haplogroup; and (b) 7 Y-STR haplotypes (DYS19, DYS391, DYS393, DYS439, DYS460, DYS461 and Y GATA A10) for the R1b1a-V88 haplogroup. Microsatellite haplotypes are represented by circles, with size proportional to their frequency in the samples and colours corresponding to their geographic regions.Haplogroup R and the ‘back to Africa’ hypothesis Figure 2.. Phylogenetic network constructed with information from (a) 10 Y-STR haplotypes (DYS19, DYS389I, DYS389II, DYS390, DYS391, DYS392, DYS393, DYS437, DYS438 and DYS439) within the R1b1-P25 haplogroup; and (b) 7 Y-STR haplotypes (DYS19, DYS391, DYS393, DYS439, DYS460, DYS461 and Y GATA A10) for the R1b1a-V88 haplogroup. Microsatellite haplotypes are represented by circles, with size proportional to their frequency in the samples and colours corresponding to their geographic regions.Haplogroup R and the ‘back to Africa’ hypothesisHaplogroup R is the most common haplogroup in European populations, and although it is usually rare in Africa, chromosomes bearing the P25-derived allele (lineage R1b1) have been reported at frequencies as high as 95% in some Central African populations.5, 6, 7, 29, 30 This haplogroup is thought to have originated in Central Asia approximately 40 000 years BP and then migrated westward into Europe, achieving its highest frequencies in the western region of this continent.39, 40 Although Balaresque et al41 proposed the hypothesis of a European spread of haplogroup R1b1b2-M269 during the Neolithic, the distribution of the M269 sub-haplogroups and their Y-STR diversities proved to be compatible with a pre-Neolithic diffusion of M269 in Europe.42, 43 The reported high frequencies of this haplogroup in Central-West Africa led to the proposal of a ‘back to Africa’ migration as the justification for the otherwise unexpected presence of this haplogroup in the region. In our sample, R1b1 was the second most frequently observed haplogroup, which was present in 17% of the sample. Ten out of the nineteen chromosomes that belong to this haplogroup present the M269-derived allele, a typical European marker and may therefore indicate recent European influx. Although European arrivals within the last five centuries have been well reported in this country, European influx was not significant in the neighbouring regions, such as Cameroon and Gabon, where this Eurasian haplogroup is rarely observed.5, 7 The remaining chromosomes presented the V88-derived allele, which was recently reported to be present in all of the typed R1b1*-P25 African chromosome.7 Furthermore, in our sample, most of the V88 chromosomes presented non-consensus alleles at the DYS385 marker, suggesting a different additional sub-lineage within this haplogroup. A phylogenetic network of R1b1 lineages based on 10 Y-STR haplotypes was constructed with samples from Cameroon and Gabon5 and from the present study (Figure 2a). A clear separation of the R1b1b2-M269 samples was observed; they clustered with the European modal haplotype, supporting their European ancestry. The non-consensus alleles observed in both studies do not show a well-defined separation from the samples with consensus alleles. However, a cluster containing haplotypes with both consensus and non-consensus alleles (representing two different lineages) and another exclusively with consensus alleles could be indicative of at least three different lineages within the R1b1-P25( × M269) haplogroup. Nonetheless, it is important to note that the data from Berniell-Lee et al5 were published before the recently discovered mutations within P25.7 Cruciani et al7 did not find any intermediate variant alleles at the locus DYS385, which were all found within haplogroup R1b1a-V88 in the present work (with only two chromosomes presenting consensus alleles). This result may indicate the presence of different sub-lineages within this haplogroup that are yet to be discovered.  The origin of the V88 lineages The origin of the V88 lineagesAlthough the recently advanced hypothesis that the V88 lineages migrated with Proto-Chadic speakers from the North Africa through the Central Sahara into the Lake Chad Basin,7 given that a high variance was found in lineages from haplogroup R1b1a in the sample from Equatorial Guinea, our results are also compatible with an origin of the V88 lineages in Central-West Africa. Assuming that Central-West Africa is in fact the place of origin for V88, the arrival of Chadic groups in the Lake Chad Basin, coming from the North, is equally likely as the alternative hypothesis of a migration mediated by the Proto-Chadic speaking people coming from East to West Africa (‘Inter-Saharan’ hypothesis), which was previously defended by Lancaster.11 According to Blench’s ‘inter-Saharan’ hypothesis, Chadic speakers originated during the eastward migration of a pastoralist Cushitic group, from the Nile towards the Lake Chad, with subsequent dispersion in different directions around the lake. The pastoralist nature of the groups involved in the dispersion of haplogroup R1b1a in Central Africa is also supported by the V88 distribution as well as by the distribution of its subclade V69 (the only one found in the African continent).7 In fact, although the origin and dispersion of these lineages seem to be much older than the beginning of the Bantu expansion, which began in this same region in Central Africa,44, 45 its frequency decreases drastically towards the South. Furthermore, in opposition to the previously suggested direction of migration,7 the presence of these lineages in North Africa could also be explained as the result of a migration of V88 carriers from South to North, possibly during the mid-Holocene. In summary, altogether, our data are compatible with an origin of the V88-derived allele in Central-West Africa and a later migration (or migrations) across the Sahara to North Africa. Assuming this origin for V88, both the ‘Trans-Saharan’ and ‘Inter-Saharan’ hypotheses for the arrival of Chadic groups in the Lake Chad Basin are equally likely, as already described in other studies,8, 46 but this model is also compatible with the dispersion of V88 from Central-West to North Africa, contrary to what was proposed by Cruciani et al.7 Nevertheless, further evidence will be required to support this hypothesis, and additional data are crucial to obtain a deeper understanding of the origin and history of this haplogroup, namely from the populations in the proposed paths of migration. González, Miguel, et al. " The genetic landscape of Equatorial Guinea and the origin and migration routes of the Y chromosome haplogroup R-V88." European Journal of Human Genetics 21.3 (2012): 324-331. |
|
|
|
Post by Admin on Sept 7, 2014 22:45:35 GMT
 Demographic change is one of the central questions in understanding human history, and strong population expansions may be linked to various events as climate changes, alteration of social structure, or technological innovations. The recent advent of next-generation sequencing technology enabled systematic analysis of the population history using the information from the whole genome with less ascertainment bias, so we can re-assess how the various factors have influenced the human population size and structure [1], [2]. Recent analyses of mitochondrial genomes revealed that the expansions of female lineages of East Asians [3] and those of Europeans [4] started before the Neolithic Era, contradictory to the hypothesis that the agricultural innovation constitutes the primary driving force of population expansions [5]. These observations prompted this study to investigate expansions of male lineages. The Y chromosome contains the longest non-recombining region (~60 Mbp, in which ~10 Mbp is unique sequence in the genome and easy to analyze) in the human genome [6], [7], making it an informative tool for reconstructing genetic relationship of human populations and paternal lineages, and dating important evolutionary and demographic events [8], [9], [10], [11]. However, the sequencing data of Y chromosomes of human populations were insufficient and biased even for those of current 1000-genome project for which coverage on Y chromosome was low (on average <1.4× in East Asian samples) [12]. According to the phylogenetic tree of Y chromosome, all the modern males could be categorized into 20 major monophyletic or paraphyletic groups (referred to as A to T) and their subclades [13], [14]. Nearly all the Y chromosomes outside Africa are derivative at the SNP M168 and belong to any of its three descendent super-haplogroups – DE, C, and F [9], [10], [15], strongly supporting the out-of-Africa theory. The time of the anatomically modern human's exodus from Africa has yielded inconsistent results ranging from 39 kya [16], 44 kya [10], 59 kya [17], 68.5 kya [18] to 57.0–74.6 kya [19].  Figure 1. Phylogenetic tree of human Y chromosome, emphasizing the three star-like expansions (Oα, Oβ, Oγ). Figure 1. Phylogenetic tree of human Y chromosome, emphasizing the three star-like expansions (Oα, Oβ, Oγ).
The tree was constructed from 78 samples sequenced in this study, together with three published East-Asian genomes and a chimpanzee genome. The branch lengths (horizontal lines) are proportional to the number of SNPs on the branch. Numbers in red indicate the coalescence time (in years, considering the variation in SNP counting, but ignoring uncertainty in mutation rate) and 95% confidence intervals of the node. For more details, see Fig. S1.Overall ~4,500 base substitutions were identified in all the samples from the whole Y chromosome, in which >4,300 SNPs that has not been publicly named before 2012 (ISOGG etc.). We designated each of these SNP a name beginning with ‘F’ (for Fudan University) (see Table S2). We obtained ~3.90 Mbp of sequences with appropriate quality (at least 1× coverage on >100 out of 110 samples, see Table S3), and identified ~3,600 SNPs in this region. A maximum parsimony phylogenetic tree of the 78 individuals with good coverage was reconstructed (Fig. 1 and Fig. S1), the topology of which is congruent with the existing tree of human Y chromosome [13], [20]. The tree contained samples from haplogroups C, D, G, J, N, O, Q, and R, and thus represented all the three super-haplogroups out of Africa – C, DE and F. In addition to the known lineages, many new downstream lineages were revealed. All the earlier divergences were found to be bifurcations, except for three star-like structures, i.e. multiple lineages branching off from a single node, were observed under Haplogroup O3a-M324, indicating strong expansion events. By using Bayesian method [21] with a constant mutation rate of 1×10−9 substitution/base/year [7], [19] (or one substitution per 256 years on 3.9 Mbp range, without considering the uncertainty of mutation rate), we calculated the date of each divergence event throughout the tree. The first divergence event out of Africa, i.e. between Haplogroup DE and the ancestor of C and F, is dated at 54.1 kya (95%CI 50.6–58.2), inside the range of previous estimations. Within the 3.9 Mbp range, only 3 SNPs were observed between the divergence events of DE/CF and C/F, indicating that DE, C, and F likely emerged subsequently in less than a thousand years. After diverged from Haplogroup C, no major split was observed in F for 18 thousand years, suggesting a strong bottleneck of F lineage. It should be noted that all the primary haplogroups (G, J, N, O, Q, and R) emerged before the last glacial maximum (LGM, ~20 kya), and most of the presently known East Eurasian clades have branched off in the late Upper Paleolithic Age (before 10 kya). All divergences on this tree before 7 kya were binary, suggesting that during the Paleolithic Age, slow population growth and bottlenecks or drift eradicated most of the ever existing clades [22].  The most surprising discovery in the tree is the three star-like expansions in Haplogroup O3-M324, i.e. under the M117 clade, the M134xM117 paragroup, and the 002611 clade. Here we denote the three star-like expansions as Oα, Oβ, and Oγ, respectively (see Discussion). Since the sample selection for high-throughput sequencing was intended for representing a wide variety of clades in East Asian populations, a star-like expansion indicates successful expansion of male lineages within a very short period (<500 years). These three clades are present with high frequency across many extant East Asian populations [23], [24] and encompass more than 40% of the present Han Chinese in total (estimated 16% for Oα, 11% for Oβ, and 14% for Oγ) [20]. It is conspicuous that roughly 300 million extant males are the patrilineal progenies of only three males in the late Neolithic Age. The expansion dates are estimated 5.4 kya for Oα, 6.5 for Oβ, and 6.8 for Oγ (Fig. 1), after the shift to intensive agriculture in North China (since 6.8 kya) [25], [26], in particular, during the Yangshao Culture (6.9–4.9 kya) in Central Yellow River Basin, Majiayao Culture (6.0–4.9 kya) in the Upper Yellow River Basin, and the Beixin (7.4–6.2 kya) – Dawenkou Culture (6.2–4.6 kya) in the Lower Yellow River Basin [27]. We therefore propose that in the late Neolithic Age, the three rapidly expanding clans established the founding patrilineal spectrum of the predecessors in East Asia. Since all the sequenced Han Chinese M117+ samples are under the Oα expansion, and M117+ subclade exists in moderate to very high frequency in many Tibeto-Burman ethnic groups [28], [29], [30], it would be of interest to know when the M117+ individuals in other ethnic groups diverged with the ones in Han Chinese, and whether they are also under the Oα expansion, in order to trace the origin and early history of Sino-Tibetan language family. Despite of the mutation rate uncertainty, we evaluate our calculation of absolute divergence time as acceptable. Firstly, our out-of-Africa date (54.1 kya) is still within the range of previous estimations (39–74.6 kya). Secondly, the out-of-Africa date is similar to the recent estimation of two great mitochondrial expansions outside Africa – M (49.6 kya) and N (58.9 kya) [43]. Thirdly, it is not contradictory to the emergence of earliest modern human fossil out of Africa (e.g. ~ 50 kya in Australia) [44]. The accumulative substitution count from the DE/CF divergence to a modern individual varies from to 168 (YCH113) to 241 (YCH198). Despite of this variation, by testing the assumption of molecular clock for the tree, the null hypothesis of a molecular clock could not be rejected (P>0.05) using PAML package v4.4 [45] with the GTR model, unlike the mitochondrial tree from complete sequences, which showed violation to the clock assumption [43]. Part of the branch length variation may come from the false negative detection of SNPs, especially on a long terminal branch; however, this effect was mostly eliminated that we chose only the sequences with good quality for time estimation, so the branch length difference for these sequences should mainly reflects the real variation, and should have little effect in time estimation.  Jiangzhai settlement model, Yangshao culture, Lintong, Shaanxi The current Y haplogroups were named according to the rule of Y Chromosome Consortium (YCC) [14]. Along with increasing clades being discovered, the present nomenclature became cumbersome in some cases, e.g. ‘R1b1b2a1a2d3a’ in the ISOGG tree 2010 (http://www.isogg.org), which is prone to frequent name changes and hard to remember. Another commonly used nomenclature such as ‘O-M117’ or ‘O-F46’ is also not suitable for a determined star-like expansion, since there are many SNPs found ancestral to the star point, while these SNPs may be found not all equivalent in the future, e.g. some individuals might be found M117+ but not belonging to the star expansion, then the name of the star point must be renewed. Therefore, here we propose a modification to the current nomenclature system: for any important star-like expansion that leads to large population (e.g. several millions) and multiple lineages (≥5) in short time as revealed by long-range sequencing (>1 Mbp was needed in order to limit the expansion within 1,000 years), a lineage name with lowercase Greek letter is applied directly after the Latin capital letter of the first-class haplogroup name. For example, the star-like expansions under M117, F46, and F11 are now named as Oα, Oβ, and Oγ, respectively, and their downstream lineages should still be named following the rule of YCC 2002, with Arabic number succeeding the Greek letter, e.g. Oα1a1. These names of the star-like expansions are not bound to any single defining SNP (e.g. M117), but to the expansion itself, i.e. the expansion names should always keep unchanged despite new side clades would be found to its upstream, in order to keep the nomenclature stable. For the currently equivalent SNPs on the branch leading to the expansion, we will know the occurring order only after vast amount of samples being genotyped for those SNPs.  Since all the Paleolithic divergences of Y chromosome lineages are binary, the three roughly contemporaneous star-like expansions revealed in this study indicate a remarkable demographic change in the late Neolithic Age. The earliest agriculture in North China emerged before 10 kya [46], however, no distinct Y chromosomal expansion could be related to this event. The three star-like expansions happened several thousand years later, thus are likely linked to middle Neolithic cultures such as Yangshao (6.9–4.9 kya) and Dawenkou Culture (6.2–4.6 kya) in the Yellow River Basin [27]. During this period, agriculture became mature and intensive, and the majority of human diet shifted from food collection into production [47], [48]. Crop harvest constituted a more stable food source than hunting and gathering, and enabled nourishing population at higher density. In addition, liberation of males from hazardous hunting might have enhanced male viability into adulthood, thus the effective population size of Y chromosome increased. Besides the progress in agriculture, changes in social structure might also contribute to the patrilineal expansion. In the middle and late phases of Yangshao and Dawenkou culture, the burial customs showed a gradual transition from an egalitarian matrilineal society into a hierarchical patrilineal one [49], [50]. Interestingly, the major maternal expansions in China shown by mitochondrial tree (among which are also several star-shaped expansions) occurred much earlier, at the late Paleolithic Age [3]. This immense non-synchrony between maternal and paternal expansion suggests a possible transition of social structure, that in the late Neolithic Age, a few paternal lineages achieved greater advantage on the existing basis of the population that started expansion since the Paleolithic Age. After the strong Neolithic expansions, the reproductive advantage of the farmers lasted for 4,000 years, until most of the gatherer-and-hunter tribes in the Yellow River Basin were absorbed by the farming societies of Huaxia, from which the Han ethnicity was formed. Although without ancient DNA proofs, we cannot yet confirm the initial expanding regions of these three clans, whether they were original in the middle or lower reach of Yellow River Valley or migrated from the vicinity, we are now at least certain that a majority of Han Chinese did derive from just a few patrilineal ancestors in the Neolithic Age. Whether each of them could be related to the legendary Emperors Yan and Huang or their tribes, is to be solved with more prudence and with the help of interdisciplinary genetic, archeological, ethnical, and documentary studies. Yan, Shi, et al. " Y Chromosomes of 40% Chinese Descend from Three Neolithic Super-Grandfathers." PloS one 9.8 (2014): e105691. |
|

