|
|
Post by Admin on Sept 11, 2014 8:07:54 GMT
 The Ashkenazi Jewish (AJ) population is a genetic isolate close to European and Middle Eastern groups, with genetic diversity patterns conducive to disease mapping. Here we report high-depth sequencing of 128 complete genomes of AJ controls. Compared with European samples, our AJ panel has 47% more novel variants per genome and is eightfold more effective at filtering benign variants out of AJ clinical genomes. Our panel improves imputation accuracy for AJ SNP arrays by 28%, and covers at least one haplotype in ≈67% of any AJ genome with long, identical-by-descent segments. Reconstruction of recent AJ history from such segments confirms a recent bottleneck of merely ≈350 individuals. Modelling of ancient histories for AJ and European populations using their joint allele frequency spectrum determines AJ to be an even admixture of European and likely Middle Eastern origins. We date the split between the two ancestral populations to ≈12–25 Kyr, suggesting a predominantly Near Eastern source for the repopulation of Europe after the Last Glacial Maximum. Comparison of tallies of variants between AJ and FL genomes (Fig. 1a; Supplementary Table 3) suggested that AJ have slightly but significantly more overall variants (+1.5%), mostly as heterozygotes. The increased AJ heterozygosity (+2.4%), in spite of the recent bottleneck, confirms previous observations (Supplementary Note 3)6, 7, 10, 19. More pertinently to the utility of a population sequencing endeavour, AJ samples have a much higher fraction (+47%) of novel variants (dbSNP135; Fig. 1a). Clinical AJ genomes will thus be screened more efficiently against the AJ reference panel. For example, an AJ genome has, on average, 36,995 novel variants (160 of which are also non-synonymous). Only 4.0% of them (3.2% for novel and non-synonymous) will be filtered out against the FL panel, whereas an AJ panel of the same size filters out 32.6% of variants (22.4%), 8.2 (7.0) times more. Using the entire AJ panel allows filtering of ≈65% of all novel variants (48%). The number of novel and non-synonymous, never-seen variants in an AJ personal genome is therefore only 83.3, making the clinical analysis of such a genome more feasible (Fig. 1b). The number of new variants discovered when sequencing each additional genome is slightly larger in our AJ cohort than in FL (Fig. 1c). However, extrapolation predicts the converse trend already for cohorts larger than n=49 samples (Fig. 1c; Supplementary Note 3; Supplementary Fig. 3), suggesting higher efficiency of the AJ cohort in cataloguing population variation.  Figure 1: Novel variants discovered in Ashkenazi Jewish and Flemish genomes. Figure 1: Novel variants discovered in Ashkenazi Jewish and Flemish genomes.
Novel variants discovered in Ashkenazi Jewish and Flemish genomes.
(a) Variant counts (all and heterozygous; left) and fraction novel (right) per genome in the Ashkenazi Jewish (AJ) and Flemish (FL) cohorts (corresponding to about ≈80% of the raw variants remaining after QC and cohort merging; Supplementary Note 2; error bars represent s.d.). (b) Efficiency of filtering all novel variants detected in an AJ personal genome, measured by counting those that remain new after filtering such a genome against either FL or AJ panels of a matched size (n=26) or our complete AJ panel (n=127). Left: all novel variants; right: non-synonymous novel variants. Error bars represent s.d. (c) The number of newly discovered segregating sites in AJ and FL versus the number of already sequenced individuals in each cohort (markers). Dashed and solid lines are expectations based on either a constant size or a bottleneck and growth model (bn/growth), respectively, fitted to each population separately (Supplementary Note 3). The inset magnifies the region (0, 10).The effective coverage of variation can also be demonstrated using identical-by-descent (IBD) segments. We detected IBD segments by using the Germline software20, with additional filtering adapted to sequencing data (Supplementary Note 4; Supplementary Fig. 4). Sharing in AJ was ≥7.9-fold more abundant than in FL or between the populations (Fig. 2a). Using the AJ panel, one can cover at least one haplotype in ≈67% of the genome of any other AJ individual with long (>3 cM) IBD segments (≈46% using segments>5 cM), compared with much poorer efficiency in Europeans (Fig. 2b; here we used the CEU panel from the 1000 Genomes project; Supplementary Note 4). These results imply that any additional, sparsely genotyped AJ sample can be effectively imputed, at least partially, along haplotypes shared with a small sequenced reference panel. Co-ancestry of copies of IBD segments is expected to be extremely recent (typically 30 or fewer generations), thus allowing only very recent mutations to be missed at the imputed genome21, 22. Whether this strategy will scale for the accurate imputation of the entire genome of an AJ proband will be resolved with the sequencing of additional genomes.  Figure 2: Utility of the AJ reference panel in IBD-based and traditional imputation. (a) The distribution, over all pairs of individuals, of the fraction of the genome shared IBD (segment lengths >3 cM) either within AJ, within FL or between AJ and FL. (b) The average fraction of a genome (in AJ and CEU) where at least one haplotype is covered by segments shared with a population-matched panel Figure 2: Utility of the AJ reference panel in IBD-based and traditional imputation. (a) The distribution, over all pairs of individuals, of the fraction of the genome shared IBD (segment lengths >3 cM) either within AJ, within FL or between AJ and FL. (b) The average fraction of a genome (in AJ and CEU) where at least one haplotype is covered by segments shared with a population-matched panelOur sequencing panel is also expected to improve the performance of traditional imputation approaches, which are known to be more accurate when the ancestries of the reference and target populations are matched23. To evaluate the quality of imputation, we divided our sequencing cohort into ‘reference’ and ‘study’ panels; in the latter, we masked all variants not genotyped on a typical SNP array. We then imputed24 the ‘study’ panel using either our ‘reference’ panel (n=50) or the larger (n=87) 1000 Genomes CEU panel18 (Supplementary Note 5; Supplementary Fig. 5). As expected, using an AJ reference panel was more accurate than using a European one, with the number of discordant genotypes 28% lower and the correlation between true and imputed dosages, r2, increasing from 97.4% to 98.2% (Supplementary Note 5; Supplementary Table 4). Using the AJ panel reduced mostly the number of false negatives (with respect to the reference genome; Supplementary Table 4); it lowered the number of wrongly imputed non-reference variants with minor allele frequency ≤1% by 2.7-fold, with the improvement remaining at 1.5–2-fold at higher frequencies (Fig. 2c; Supplementary Fig. 6). This improvement in imputation quality likely reflects both the increased segmental sharing in AJ as well as the large number of AJ-specific alleles. These results motivate using a population-matched, rather than a merely continent-matched, reference panel, even for the closely related AJ and European populations. Our sequencing data also enables detailed reconstruction of AJ and European population histories. Allele frequency spectra (AFS) are attractive conduits for such an analysis, especially in deeply sequenced cohorts. The AFS of both AJ and FL (Fig. 3a) reject a constant-size population model, which has previously been ruled out across multiple human populations25. The two spectra are similar, with AJ showing a slight excess of doubletons. These spectra each fit well to similar models of ancient history, comprising an ancient bottleneck (≈60–86 Kyr) followed by slow exponential growth (Supplementary Note 6; Supplementary Table 5; Supplementary Fig. 7; Supplementary Fig. 8). The joint (AJ–FL) AFS reveals correlated allele counts (Fig. 3b), indicating gene flow between the populations or very recent divergence (Supplementary Note 6). Yet, correlation is not as strong as it would have been had the AJ–FL combined sample been panmictic (Fig. 3b; FST=0.016; Supplementary Note 6). The normalized AFS of population-specific variants (Fig. 3a, inset) is noticeably different between AJ and FL, with higher allele frequencies in AJ. There were overall 14% more population-specific variants in AJ (Supplementary Note 6; Supplementary Figs 9 and 10), pointing to asymmetric gene flow from Europeans into the ancestral population of AJ.  Figure 3: The AFS and the lengths of shared segments. (a) The (normalized) minor allele frequency spectrum in AJ and FL, shown as counts in subsets of n=25 genomes in each cohort. The green line corresponds to the expectation in a constant-size population (Wright–Fisher), and bars represent deviations in AJ and FL. The inset shows the spectra of alleles private to each population. (b) A heat map of the joint (minor) allele frequency spectrum of AJ and FL (lower left triangle) compared with the expected joint AFS, had population labels been random (upper right triangle)33. (c) The average fraction of the genome found in shared segments versus the segment length (AJ only; circles), along with the best fit to a recent bottleneck and growth model (solid blue line; Fig. 4) and the expectation in a constant-size population with the same total sharing (dashed green line). Figure 3: The AFS and the lengths of shared segments. (a) The (normalized) minor allele frequency spectrum in AJ and FL, shown as counts in subsets of n=25 genomes in each cohort. The green line corresponds to the expectation in a constant-size population (Wright–Fisher), and bars represent deviations in AJ and FL. The inset shows the spectra of alleles private to each population. (b) A heat map of the joint (minor) allele frequency spectrum of AJ and FL (lower left triangle) compared with the expected joint AFS, had population labels been random (upper right triangle)33. (c) The average fraction of the genome found in shared segments versus the segment length (AJ only; circles), along with the best fit to a recent bottleneck and growth model (solid blue line; Fig. 4) and the expectation in a constant-size population with the same total sharing (dashed green line).We next turned to inferring an explicit model for the demographic history of AJ and Europeans. Since the allele frequency spectrum, in particular for our sample size, may not be sensitive to recent demographic events, we first reconstructed the very recent AJ history by examining long IBD segments5, 12, 14, 21, which carry information on recent co-ancestry (last ≈50 generations). We used the lengths of shared segments (Fig. 3c) to infer the parameters of a recent AJ bottleneck (effective size 250–420; 25–32 generations ago) followed by rapid exponential expansion (rate per generation 16–53%; Fig. 4, bottom), confirming previous analyses conducted on lower throughput data (Supplementary Note 4; Supplementary Table 6; Supplementary Fig. 11)12, 14.  Figure 4: A reconstruction of the AJ and FL demographic history. The upper part of the diagram shows the reconstruction of the ancient history by fitting the joint AFS (Fig. 3b) using ∂a∂i26 and using a mutation rate of 1.44 × 10−8 per generation per bp. The lower diagram shows the recent AJ history, reconstructed by fitting the IBD length decay pattern (Fig. 3c). The wide arrow represents an admixture event; all effective population sizes (horizontal arrows) are in number of diploid individuals; all times were computed assuming 25 years per generation. Confidence intervals are provided in Supplementary Tables 6 and 7. Figure 4: A reconstruction of the AJ and FL demographic history. The upper part of the diagram shows the reconstruction of the ancient history by fitting the joint AFS (Fig. 3b) using ∂a∂i26 and using a mutation rate of 1.44 × 10−8 per generation per bp. The lower diagram shows the recent AJ history, reconstructed by fitting the IBD length decay pattern (Fig. 3c). The wide arrow represents an admixture event; all effective population sizes (horizontal arrows) are in number of diploid individuals; all times were computed assuming 25 years per generation. Confidence intervals are provided in Supplementary Tables 6 and 7.Given the model for the recent AJ history, we inferred the parameters of a model for the ancient history of AJ and FL using an existing method (∂a∂i26) based on the joint frequency spectrum (Supplementary Note 6; Supplementary Data 3). Confidence intervals were computed using parametric bootstrap26 (Supplementary Note 6), but we did not integrate over the uncertainty in the mutation rate (see the next paragraph). According to the resulting model (Fig. 4, top; Supplementary Table 7; Supplementary Fig. 12), contemporary AJ formed 600–800 years (close to the time of the AJ bottleneck) as the fusion of two ancestral populations. One ancestral population, consistent with being the ancestors of the FL samples, contributed 46–50% of the AJ gene pool. We call that population ancestral European and the other ancestral Middle Eastern. The ancestral European population went through a founding bottleneck (effective size 3,500–3,900) when diverging from ancestral Middle Easterners. We date this event to 20.4–22.1 Kyr, at around the time of the Last Glacial Maximum and preceding the Neolithic revolution (27; see Supplementary Note 6 and below for discussion). The ancestors of both populations underwent a bottleneck (3,600–4,100 founders) at 85–94 Kyr, likely corresponding to an Out-of-Africa event28. Previous explicit demographic models using genome-wide SNP arrays or low-pass sequencing data time-stamped a European bottleneck at ≈40–80 Kyr (recalibrated to the lower mutation rate estimate; Supplementary Note 6), with even the lowest estimates26, 33, 34 being higher than our point estimate of ≈21 Kyr. However, no previous study has employed deeply sequenced genomes of (partial) Middle Eastern ancestry; in addition, previous studies usually modelled the European founder event simultaneously with the divergence from East Asian populations. As modern humans had colonized Europe already by ≈40–45 Kyr35, our results (across all estimates of the mutation rate) support genetic discontinuity between that (hunter–gatherer) population and contemporary Europeans. A Middle Eastern European divergence time around ≈21 Kyr would also suggest (i) a near Eastern source for the repopulation of Europe at the end of the Last Glacial Maximum27, 36 and (ii) that migration from the Middle East to Europe largely preceded the Neolithic revolution, suggesting that Neolithic population movements were largely within Europe37, 38, 39, 40, 41, 42. These interpretations, however, strongly depend on the mutation rate: taking into account the uncertainty in the mutation rate, our divergence time estimate is between ≈12–25 Kyr, which can be reconciled with Neolithic migrations originating in the Middle East (Supplementary Note 6). Carmi, S. et al. Sequencing an Ashkenazi reference panel supports population-targeted personal genomics and illuminates Jewish and European origins. Nat. Commun. 5:4835 doi: 10.1038/ncomms5835 (2014). |
|
|
|
Post by Admin on Sept 22, 2014 22:53:26 GMT
 At the beginning of the first millennium BC the following native tribes could be distinguished on the Italian territory: the Ligures, on the coast that bears their name, in the northern Apennine valleys, part of the pre-alpine valleys and the western Po Valley; the Sicani, in the interior of Sicily; and the Itali, in present-day Calabria (from whom comes the name ‘Italy’, which was to be extended to all the territory of the peninsula). Besides the already mentioned Terramare tribe, on the southern edge of the Po Valley, and the Villanovans, probably from Eastern Europe who settled throughout Central Italy, there were also the Umbrians to the east of the upper basin of the Tiber. The Veneti, who occupied the territory that still bears their name, originally came from Illyria as did the Messapii (now modern Salento or South Apulia) and Iapyges, who settled in present-day Puglia (Apulia) [5]. Many other populations of Central-Southern Italy were created by the mixing of local and foreign elements dating back to the previous millennium; it is the case of the Sabines and Latini who settled in Lazio together with Falisci, Aequi, Volsci, Hernici and Ausones. The interior of Abruzzo was dominated by the Vestini, Paeligni and Marsi, while the central Adriatic coast was populated by Picentes, Marrucini and Frentani. The Apennine area of Molise and Basilicata was peopled by the Samnites and Lucanians. In Calabria and Sicily there were also the Bruttii and Siculi. The Phoenician colonization of the coasts of the Western Mediterranean were mainly limited in Italy to Sardinia and western Sicily and preceded that of the Greeks. It was followed by Punic settlements (Trapani, Palermo, Cagliari) linked to the ancient Phoenician colony of Carthage. At the time of the Roman Empire, at least two non-Indo-European populations still inhabited Italy, namely, the Ligures, in the northwestern area, and the Etruscans with settlements located in areas far from the Etruria (Tuscany and High Latium), such as the Po Plain and the coast of Campania. At the same time, Sardinia experienced the flourishing of a non-Indo-European Nuragic civilization and, then, the Phoenician colonization. Genetics alone cannot disentangle the extremely complex demography of Italy through history. Some demographic movements have however left signals on uniparental and nuclear markers. Most of the genetic studies targeted local, e.g. [7], or regional, e.g. [8]–[11], Italian populations.  Figure 1. Map showing the location of the samples analyzed in the present study and those collected from the literature (see Table 1). Pie charts on the left display the distribution of mtDNA haplogroup frequencies, and those on the right the Y-chromosome haplogroup frequencies. Figure 1. Map showing the location of the samples analyzed in the present study and those collected from the literature (see Table 1). Pie charts on the left display the distribution of mtDNA haplogroup frequencies, and those on the right the Y-chromosome haplogroup frequencies.For the Y-chromosome, some attempts have been undertaken to analyze Italian variation to a more general scale [12]–[14]. Many studies have analyzed specific haplogroups in the Y-chromosomes, e.g. [15], [16], or the mtDNA, e.g. [8], [9]. In general, the different studies indicate that the genetic structure of the present Italian population seems to reflect, at least in part, the ethnic stratification of pre-Roman times [14]. Studies carried out in the past appear to show a major North–South cline consistent with archaeological estimates of two distinct processes: the first colonization of the area during the Paleolithic period and the subsequent Neolithic expansion from the Middle East after the last glacial [14]. There is some correspondence between patterns of variation at the Y-chromosome and geography. Thus, northern Italy shows similar frequencies as the haplogroups of Central Europe, with prevalence of the western R1-M173 haplogroup compare to the eastern I-M170. In the North, E3b1-M35 and J2-M172 show low frequencies but are more prevalent in the South, which has been interpreted to be a signal of the gene flow coming from Central European Neolithic farmers [17]. R1a1-M17 is rather rare, both in the North, where it probably originates from eastern Europe, and in the South, of possible Greek provenience [17]. Occurrence of J2-M172 Y-chromosomes in Tuscany has been related to the Etruscan heritage of the region (see [17]). The two Italian major islands, Sicily and Sardinia, show a different demographic history. The Y-chromosome variability of Sicily shares a common history with that of southern Italy, enriched by an additional Arab contribution, but also North African and Greek influences [18]. On the other hand, Sardinia has been considered to be a genetic outlier within Europe showing clear signals of founder effects; some scholars suggest that its peoples could be of ancient Iberian origin [19]; recent genetic studies point to genetic contribution coming from southern France [20]. On the other hand, mitochondrial DNA studies show that Italy does not differ too much from other European populations; however, some populations have the same peculiarities and preserve signals of the ancient past demographic event, such as the Tuscans [8], [9], or the Ladins [7], [21], [22]. Recently, patterns of variation observed in haplogroup U5b3 demonstrated for the first time the existence of a North Italian pre-historical human refuge from the hostile Central European regions covered by the ice of the Last Glacial Maximum period [20]; this area, as was also the Franco-Cantabrian region [23]–[26], served as a region of European repopulation during the beginning of the Holocene. 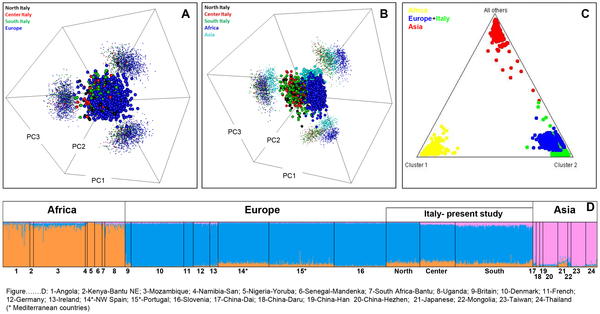 Figure 2. Analysis of AIMs in Italian populations versus other continental population groups. (A) PCA of Italian populations divided into the main regions North, Center and South (as analyzed in the present study) and other European populations; (B) the same Italian populations plus sub-Saharan African, and Asian populations; (C) triangle plot as obtained using STRUCTURE analysis of Italian, European, sub-Saharan, and Asian populations; (D) bar plot of ancestral membership values as obtained using STRUCTURE analysis of the same populations used in (C). Population codes: 1: Angola; 2: Kenya-Bantu NE; 3: Mozambique; 4: Namibia-San; 5: Nigeria-Yoruba; 6: Senegal-Mandenka; 7: South Africa-Bantu; 8: Uganda; 9: Britain; 10: Denmark; 11: French; 12: Germany; 13: Ireland; 14*: NW Spain; 15*: Portugal; 16: Slovenia; 17: China-Dai; 18: China-Daru; 19: China-Han; 20: China-Hezhen; 21: Japanese; 22: Mongolia; 23: Taiwan; 24: Thailand. Genotypes were downloaded using the method in [43], [83] and belong to the CEPH panel. An asterisk indicates Mediterranean populations. Figure 2. Analysis of AIMs in Italian populations versus other continental population groups. (A) PCA of Italian populations divided into the main regions North, Center and South (as analyzed in the present study) and other European populations; (B) the same Italian populations plus sub-Saharan African, and Asian populations; (C) triangle plot as obtained using STRUCTURE analysis of Italian, European, sub-Saharan, and Asian populations; (D) bar plot of ancestral membership values as obtained using STRUCTURE analysis of the same populations used in (C). Population codes: 1: Angola; 2: Kenya-Bantu NE; 3: Mozambique; 4: Namibia-San; 5: Nigeria-Yoruba; 6: Senegal-Mandenka; 7: South Africa-Bantu; 8: Uganda; 9: Britain; 10: Denmark; 11: French; 12: Germany; 13: Ireland; 14*: NW Spain; 15*: Portugal; 16: Slovenia; 17: China-Dai; 18: China-Daru; 19: China-Han; 20: China-Hezhen; 21: Japanese; 22: Mongolia; 23: Taiwan; 24: Thailand. Genotypes were downloaded using the method in [43], [83] and belong to the CEPH panel. An asterisk indicates Mediterranean populations.The mtDNA haplogroup make-up of Italy as observed in our samples fits well with expectations in a typical European population. Thus, most of the Italian mtDNAs (~89%) could be attributed to European haplogroups H (~40%), I (~3%), J (~9%), T (~11%), U (~20%; U minus U6), V (~3%), X (~2%) and W (~1%); Figure 1. There are however important differences in haplogroup frequencies when examining them by main geographical regions. Thus, for instance, haplogroup H is 59% in the North, 46% in the Center, and decays to ~33% in the South; moreover, these regional differences are statistically significant: North vs South (Pearson's chi-square, unadjusted-P value<0.00003), and Center vs South (Pearson's chi-square, unadjusted-P value<0.03724). 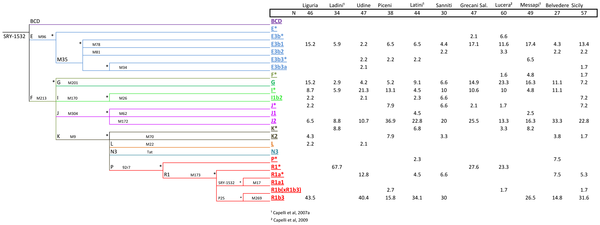 Figure 3. Phylogeny of Y-chromosome SNPs and haplogroup frequencies in different Italian populations. Figure 3. Phylogeny of Y-chromosome SNPs and haplogroup frequencies in different Italian populations.Mitochondrial DNA haplotypes of African origin are mainly represented by haplogroups M1 (0.3%), U6 (0.8%) and L (1.2%); from here onwards, L will be used to refer to all mtDNA lineages, excluding the non-African branches N and M [60], [61]. A total of 282 Y-chromosomes were analyzed for a set of Y-SNPs and were classified into 22 different haplogroups (Figure 3). Two haplogroups were not found, even though markers defining these clades were tested: N3 and R1a1. Five haplogroups represented 76.71% of the total chromosomes: R1b3, J2, I(xI1b2), E3b1 and G. The frequencies averaged across populations were 26%, 21.2%, 10.2%, 9.9% and 9.2%, respectively. The remaining haplogroups sum to 23.2% in the total sample, and never above 4% in single population samples. R1b3 frequency was found to be higher in the northern part of the country, while the Y-chromosome haplogroups G and E3b1, J2 and I(xI1b2)frequencies were higher in the south and in the central part of the country, respectively (Figure 1). Regional differences are substantially higher in the Y-chromosome than in the mtDNA. Thus, for instance, haplogroup R in the Y-chromosome was 54% in the North, 18% in the Center, and 31% in the South. Frequency differences were statistically significant between North vs Center (Pearson's chi-square, unadjusted-P value = 0.0014), and North vs South (Pearson's chi-square, unadjusted-P value<0.00004). Haplogroup J2 also revealed important regional differences; it added to 9% in the North, 37% in the Center, and 22% in the South, with statistically significant differences between the North vs Center (Pearson's chi-square, unadjusted-P value<0.00002), North vs South (Pearson's chi-square, unadjusted-P value<0.00148), and in the limit of significance Center vs South (Pearson's chi-square, unadjusted-P value<0.049). A panel of 52 AIMs was genotyped in 435 Italian individuals in order to estimate the proportion of ancestry from a three-way differentiation: sub-Saharan Africa, Europe and Asia. Structure analyses allowed us to infer membership proportions in population samples, and these proportions can be graphically displayed, as in Figure 2. This analysis indicated that Italians have a basal proportion of sub-Saharan ancestry that is higher (9.2%, on average) than other central or northern European populations (1.5%, on average). The amount of African ancestry in Italians is however more comparable to (but slightly higher than) the average in other Mediterranean countries (7.1%). Figure 2 shows in a triangle plot the relationships of Italians compared to other European, African and Asian populations. PCA observations confirmed the results from Structure analysis, clustering Italian profiles tightly with other European ones. Thus, PCA indicated that North, Central and South Italy do not show differences between them, nor from other European populations (Figure 2). PCA also indicated clear-cut differences between Italians, Africans and Asians (Figure 2).  Figure 4. Haplogroup frequencies of Ladins, Grecani Salentini and Lucera compared to the rest of the Italian populations analyzed in the present study. Figure 4. Haplogroup frequencies of Ladins, Grecani Salentini and Lucera compared to the rest of the Italian populations analyzed in the present study.The differences between Ladin and other populations were more evident when examining haplogroup frequency patterns (Figure 4). The frequency of haplogroup H (58%) was above the frequency of H in North Italy (55%), and was extremely high (58%) compared to the average for Italy (38%) (Pearson's Chi-square test, P-value = 0.0005). While haplogroup U was found to have approximately the same frequency as other Italian populations, haplogroup T was 5% compared to 12% in Italy generally (7% in the North). Other differences were apparent, but sample sizes were relatively low to yield significant statistical differences. Differences are more important when examining Y-chromosome haplogroup frequencies. R1b3 reached 52% in Ladin populations but only 31% in the general population, and also in the North (Pearson's Chi-square test, P-value = 0.0087); Figure 4. More remarkable are the differences when considering the remaining R1b lineages, that is, R1b(xR1b3), which account for 15% of the lineages in Ladins, but only for 1% in the general population (Pearson's Chi-square test, P-value = 0.0001). Other haplogroups showed substantial haplogroup differences (e.g. J2) but the sample size was again too small. Due to the availability of data for mtDNA in several Ladin communities, we were able to carry out an AMOVA analysis in order to investigate the level of population stratification in these communities. The data indicated that among-population variance is 1.09%, a value that is therefore higher than the average for the Italian Peninsula (0.79%; Table 4). The North African historical legacy in South ItalyWe sampled 60 individuals from Lucera. This population sample showed diversity values that fell within the average of a typical Italian population, regarding the mtDNA (Table 1) and the Y-chromosome (Table 2). Additionally, at the level of haplogroup frequencies, Lucera matched well with other Italian populations (Figure 4). There are two mtDNA haplogroups, namely U6 and M1 that can be considered to be of North African origin and could therefore be used to signal the documented historical input of this African region into Lucera. In our full set of samples, we observed five U6 haplotypes belonging to sub-haplogroups U6a, U6a2, and U6a4. Only one of these haplotypes was observed in Lucera. However, the other three U6 haplotypes were observed in the vicinity of the population of South Apulia, and another at the tip of the Peninsula (Calabria). Regarding M1 haplotypes, we observed only two carriers in our samples sharing the same HVS-I haplotype; both were found in Trapani (West Sicily). Therefore, while South Italy shows evidence of having female introgression from North Africa, this African influence seems not to be particularly centered in the Lucera. In the Y-chromosome, we did not observe any signal of North African introgression; at least, no more than for other regions of Italy (perhaps with the exception of Sicily [31]). This again contrasts with the results of previous studies based on the Y-chromosome (but at higher or different level of phylogenetic resolution involving the genotyping of African minor sub-lineages) where signals of North African influence were observed at this latitude of the Peninsula [31]. DiscussionThe Grecani Salentini also showed signatures of genetic isolation when compared to other Italian populations, but the differences are not as marked as observed for the Ladins. The differences with respect to neighboring Italian populations were not evident when observing individual haplotypes (as occurs with the Ladins), but were clearer when considering haplogroup frequencies (Figure 4). Larger sample sizes are needed in order to gather more signatures about the demographic past of this population. Thus, the Ladins show a more distinctive pattern than the Grecani Salentini, which is to be expected given that not only is the Ladin population a linguistic isolate, but also that these communities are confined to isolated geographical areas of the Alps. Apart from the regional and local genetic differences observed in Italy, it is also worth examining global genetic patterns along the length of continental Italy. Geographical clines of Y-chromosome haplogroups in Europe have been previously reported in the literature [13]; these patterns have found support in archaeological and linguistic evidence. In the Italian peninsula, the Y-chromosome variation also shows a clinal pattern along the North–South axis; the Mesolithic haplogroup R1*(xR1a1) shows higher frequency in the North while the Neolithic haplogroup J2-M172 is superposed to this Mesolithic strata with frequency patterns running in the opposite direction [14], [69]. The results of the present study agreed with these earlier findings. Thus, for instance, R1b3 reached 31% in the North, 16% in the Center, and 14% in the South. Frequency of Y-chromosome haplogroup J2 was found to be 9% in the North, 37% in the Center, and 22% in the South (average in Italy: 14.5%). Haplogroup J2 is widely believed to be associated with the spread of agriculture from Mesopotamia. The main spread of J2 into the Mediterranean area is thought to have coincided with the expansion of agricultural populations during the Neolithic period. As reported by Di Giacomo et al. [12], haplogroup J “…constitutes not only the signature of a single wave-of-advance from the Levant but, to a greater extent, also of the expansion of the Greek world, with an accompanying novel quota of genetic variation produced during its demographic growth…”; also that “…in the central and west Mediterranean, the entry of J chromosomes may have occurred mainly by sea, i.e., in the south–east of both Spain and Italy…”. J2-M12 is almost totally represented by its sublineage J2-M102, which shows frequency peaks in both the southern Balkans and north-central Italy (14%; [13]). J2-M67 is most frequent in the Caucasus, and J2-M92 indicates affinity between Anatolia and southern Italy (21.6%; [13]). For the J1-M170 clade, the peaks of J1-M267 are in the Levant and in northern Africa, and it is closely associated to the diffusion of the Arab people, dropping abruptly outside of this area (including Anatolia and the Iberian peninsula), even if it shows an appreciable percentage in Sicily [70]. In a recent study, Pala et al. [71] confirmed that mtDNA haplogroups J and T and their major sub-clades (J1 and J2, T1 and T2) most likely arose in the Near East at the time of the first settlement by modern humans and the LGM. These haplogroups started to spread from the Near East into Europe immediately after the peak of the last glaciation, about 19 kya ago, with a major expansions in Europe in the Late Glacial period, about 16–12 kya ago, thus indicating that many of the Neolithic expansions from southern Europe into Central Europe and the Mediterranean might have been indigenous dispersal of these lineages. Latitudinal clinal frequency patterns are also observed for the mtDNA haplogroups mirroring those of the Y-chromosome. As reported by Richards et al. [38], haplogroups H, K, T*, T2, W, and X are the major contributors to the Late Upper Paleolithic, and the central-Mediterranean region has the greatest Middle Upper Paleolithic component outside the Caucasus. In agreement with the Y-chromosome, we observed that all these Paleolithic haplogroups together add to approximately 70.3% in the North, 60.8% in the Center, and 54% in the South of Italy. The opposite pattern was observed for the main mtDNA Neolithic component, represented by haplogroups J and T1, which accounted for 5.8% in the North, 10.3% in the Center, and 14.1% in the South (Italian average: 10.5%). As early as 1934, [72], Vere Gordon Childe suggested that the indigenous communities of hunters and gatherers of the Mesolithic European cultures were replaced by communities of farmers migrating to the North from the Middle East, a process that lasted for several generations. The first stream of emigration followed the route along the continental Balkan Peninsula and the Danube, while another, slightly later, emigration spread along the coasts of the Mediterranean Sea from East to West. The latter path would fit well with the distribution of other Neolithic cultural features, such as the so-called Cardium Pottery (or Cardial Ware) [73], the ceramic decorative style that better defines the Neolithic culture. This culture entered from Greece towards the South-Center of Italy through the Adriatic Sea, carried by the same farmers that introduced, for instance, Y-chromosome haplogroup J2 at about the same frequency in Central and South Italy, but with lower introgression into the North; from here followed further Mediterranean expansions towards Iberia. The sub-clade E3b1 (probably originating in eastern Africa) has a wide distribution in sub-Saharan Africa, Middle East and Europe. This haplogroup reaches a frequency of 8% in the North and Center and slightly higher in the South of Italy, 11% (Figure 1). It has also been argued that the European distribution of E3b1 is compatible with the Neolithic demic diffusion of agriculture [15]; thus, two sub-clades, E3b1a- M78 and E3b1c-M123 present a higher occurrence in Anatolia, the Balkans and the Italian peninsula. Another sub-clade, E3b1b-M81 is associated with the Berber populations and is commonly found in regions that have had historical gene flow with Northern Africa, such as the Iberian peninsula [74], [75]–[76]–[78], including the Canary Islands [75], and Sicily [70], [79]; the absence of microsatellite variation suggests a very recent arrival from North Africa [80]. If we assume that all E3b1 represents the only Y-chromosome continental African contribution to Italy and L and U6 lineages the continental African mtDNA component, the African component in Italy is higher for the Y-chromosome (8–11%) than for mtDNA (1–2%). The origin of sub-Saharan African mtDNAs in Europe (including Italian samples) has been recently investigated by Cerezo et al. [81]; the results indicate that a significant proportion of these lineages could have arrived in Italy more than 10,000 years ago; therefore, their presence in Europe does not necessarily date to the time of the Roman Empire, the Atlantic slave trade or to modern migration. In addition, the Northern African influence in the Italian Peninsula is evidenced by the presence of Northern African Y chromosome haplogroups (E1-M78) in three geographically close samples across the southern Apennine mountains: East Campania, Northwest Apulia and Lucera [31]. The Lucera sample analyzed in the present study did not however show a higher impact from North Africa than for other areas from southern Italy [31]. Finally, in agreement with uniparental markers, analysis of AIMs as carried out in the present study indicated that Italy shows a very minor sub-Saharan African component that is, however, slightly higher than non-Mediterranean Europe. This agrees with the recent findings of Cerezo et al. [82] based on the analysis of entire mtDNA genomes pointing to the arrival in ancient and historical times of sub-Saharan African people to the Mediterranean Europe, followed by admixture. Brisighelli, Francesca, et al. " Uniparental markers of contemporary Italian population reveals details on its pre-Roman heritage." PloS one 7.12 (2012): e50794. |
|
|
|
Post by Admin on Oct 9, 2014 21:40:45 GMT
 People from the Roma community sit on a bus after expulsion from their camp on June 5, 2013 in Lille, northern France. Population samples of 69 Gypsy individuals belonging to the major subdivision of the Polish Roma (Polska Roma) were studied. The samples were collected in the West of the country, in the urban areas of Zielona Góra and Nowa Sól. All individuals were maternally and paternally unrelated and originated from the area considered for this study. Appropriate informed consent was obtained from all participants. Total genomic DNA was extracted from hairs by means of cell lysis in the presence of proteinase K and 1% SDS, followed by phenol/chloroform extractions. RFLP typing was performed by restriction endonuclease analysis of PCR-amplified mtDNA fragments using the same primer pairs and amplification conditions as described elsewhere (Torroni et al. 1996; Finnil et al. 2000) (Table 1). The samples were typed for a restricted set of RFLPs that were diagnostic of all major Eurasian clusters, on the basis of the hierarchical mtDNA RFLP scheme (Macaulay et al. 1999; Richards et al. 2000; Yao et al. 2002; Malyarchuk et al. 2003). A total of eight haplogroups were identified, out of which three - H, J1 and U3 - accounted for 71% of all individuals. The remaining five haplogroups – namely, M*, I, W, X and K – occurred at lower frequencies (<10%). Among these haplogroups, haplogroup M* was found at a frequency of 5.8%, although this haplogroup has been found previously at high frequency in different Romani populations, accounting for 26.5% of the total sample studied (Gresham et al. 2001) (Table 3). The ancestral HVS I motif of haplogroup M* in the Roma is 16129-16223-16291-16298. Previously, Gresham et al. (2001) identified this cluster of HVS I sequences as belonging to haplogroup M5 described in Indians by Bamshad et al. (2001). Meanwhile, the exact position of the Romani-specific M-lineages on the evolutionary tree remains unclear, despite the current progress with mtDNA classification in populations from the Indian subcontinent (Metspalu et al. 2004; Palanichamy et al. 2004; Rajkumar et al. 2005).  Among four Polish Roma individuals characterized by M*-haplotypes we found four HVS I/II sequence types differing by nucleotide substitutions at positions 16234 and 146, as well as by an additional point insertion at position 309 (Table 2). Since the combined HVS I /II sequencing approach did not provide any useful information on the cluster-specific mutations in the HVS II region, we performed a search of diagnostic mutations by means of complete mtDNA sequencing. This study allowed us to reveal a large number of mutations distinguishing the M*-lineage from the rCRS-sequence (Figure 1). Comparison of the Romani M*-lineage with the Indian M5-sequence (Bhovi individual Bho134 from the study by Rajkumar et al. 2005) demonstrated that the haplogroup M5 is characterized by three transitions in the coding region, at sites 12477, 3921 and 709. Therefore, the results obtained clearly indicate that the Romani M*-lineage belongs to the Indian-specific haplogroup M5.  Haplogroup H is one of the most frequent mitochondrial haplogroups in the Roma (Gresham et al. 2001). In the Polish Roma it was found at a frequency of 16%. This haplogroup is also the most frequent haplogroup in Europe and is characterized by a considerable branching substructure with several large subclusters (Finniläet al. 2001; Herrnstadt et al. 2002; Achilli et al. 2004; Loogväli et al. 2004). Among the Roma, only one H-haplotype (16261-16304) is widespread in different Romani populations of Europe, but it has the highest frequency (11%) in the Vlax Roma. In Europeans this haplotype is very rare, being found only in several individuals from Belorussian and Bosnian populations (Belyaeva et al. 2003; Malyarchuk et al. 2003). Interestingly, haplotype 16261-16304 has still not been found in populations from India and Pakistan (Kivisild et al. 1999; 2003, Quintana-Murci et al. 2004). Haplogroup U3 is also one of the most frequent haplogroups in the Roma, but its highest frequencies were found in the Spanish, Lithuanian and Polish Roma (Table 3). Diversity of the haplogroup U3 in the Roma is reduced mainly to a single haplotype 16343 that is a root haplotype for haplogroup U3. Note that this haplotype is also present in many populations from Europe and the Middle East (Richards et al. 2000). Haplogroup J is characterized by a very high frequency in the Polish Roma (18.8%). This haplogroup has been also found frequently in other Roma populations (Table 3), but it is noteworthy that haplogroup J appears to be very diverse in the Bulgarian Roma - 8 out of 11 HVS I sequence types found in European Roma were observed in individuals belonging to the Balkan and Vlax groups (Gresham et al. 2001). In contrast, the Polish Roma are characterized by a marked “founder” effect, because all of their 13 J-individuals have a single HVS I/II sequence type, bearing transitions at positions 16235 and 16271 in the HVS I region and belonging to the subhaplogroup J1. The haplotype with the HVS I motif 16069-16126-16145-16222-16235-16261-16271 is very rare in European Roma populations, being found only in the Spanish Roma (one occurrence). Among Europeans, this haplotype has been revealed only in French (0.5%; Dubut et al. 2004) and Czech (2.3%; Vanecek et al. 2004) populations. A similar haplotype, lacking only the 16271 transition, has been revealed in a single individual from Bulgaria (Kalaidjii North population) (Gresham et al. 2001). It is important that this haplotype has also been found in Baluch and Brahui populations from Southwestern Pakistan at frequencies of 5% and 7.9%, respectively (Quintana-Murci et al. 2004). Derived haplotypes, with an additional transition at position 16189, were also described in populations from Syria and Turkey (Richards et al. 2000). Therefore, one may suggest that J1-haplotypes characterized by mutation at position 16235 might have been characteristic of the ancestral Romani population. HVS I sequence type 16145-16223 is another mtDNA haplotype frequent in the Polish Roma (8.7%) but absent in other Romani populations. The results of RFLP typing have shown that this haplotype belongs to haplogroup W, so the absence of the W-specific mutation at position 16292 may be due to a back mutation. An analysis of the database of Richards et al. (2000) shows that position 16292 appears to be stable within the haplogroup W members. Nevertheless, recent data has indicated several cases of a back mutation at this position within haplogroup W (Palanichamy et al. 2004). Population screening of haplotype 16145-16223 in published data sets from different Eurasian populations has shown a lack of this haplotype. Similar W-haplotypes characterised by a transition at position 16145 were found only in some populations from India (Gujarati) and Pakistan (Sindhi) (according to data of Quintana-Murci et al. 2004). Another relatively frequent haplotype among the Polish Roma (4.3%) is the HVS I sequence 16224-16234-16311 belonging to haplogroup K. This haplotype has not been found up to now in any Romani population (Gresham et al. 2001). Population screening has shown that haplotype 16224-16234-16311 is rare among European and Near Eastern populations but is very frequent (24%) in Ashkenazi Jewish populations (Behar et al. 2004). This haplotype has not been found in Poles (Malyarchuk et al. 2002) but is relatively frequent in the Polish Ashkenazi (7.3%) (Behar et al. 2004). The remaining haplotypes (Tables 2 and 4) found in the Polish Roma belong to haplogroups H, I, and X. These haplotypes have been observed in different populations of Eurasia (Table 4). Among haplogroup X lineages found in the Roma one specific subcluster defined by a transversion at position 16189 is interesting, since it appears to be non-typical for European populations. This subcluster was not found in the Lithuanian and Spanish Roma, and is rare in the Polish Roma (1.4%), but is common in the Bulgarian Roma groups being found at frequencies of 5.6 and 8.5% in the Vlax and Balkan Roma, respectively (Gresham et al. 2001). Recent population screening of haplogroup X diversity in Eurasia and North Africa has shown that this subcluster (within subgroup X2e) is only found in several individuals from southern Europe, but the Roma-specific branch defined by the 16126 transition is virtually absent in Eurasian populations (Reidla et al. 2003). The same is true for other X-haplotypes defined by a mutation at position 16241, which were found in the Roma populations (Gresham et al. 2001). These haplotypes have been described in only two Russian individuals from South Russia (Malyarchuk et al. 2002). In general, only the presence of haplogroup M5 in different Roma populations clearly points to the Asian origin of this founding Romani lineage. Note that according to Y-chromosome variation data, the paternal lineage of Asian origin (similar to maternal M5) identified in all Romani populations is haplogroup H1, defined by the M82 marker (Gresham et al. 2001). Thus, the high frequency of several West Eurasian mtDNA haplotypes that are rare or absent in European populations (such as J1, H (261–304), and W) but present in the Polish Roma may be an indication of the effects of genetic drift acting on this population (Table 4). ConclusionsThe previous analysis of the relevance of different criteria (cultural, historical, linguistic, geographic) to the genetic structure of maternal DNA lineages in the Roma performed by Gresham et al. (2001) revealed a complex pattern. However, combined analysis of both maternal and paternal lineages allows for the suggestion that classification based on the history of migrations can result in the most highly significant intergroup differences (Gresham et al. 2001). It has been indicated that the current genetic structure of the European Roma resulted mainly from early splits and divergent migration routes within Europe (Gresham et al. 2001). Genetic drift and different levels and sources of admixture are thought to be two general processes explaining the pattern of observed differentiation of the Roma populations. Our data also indicate that the effects of genetic drift are likely to account for the differences in the distribution of mtDNA lineages in different Romani populations. However, it is difficult to explain the uneven frequency of haplogroup U3 in the Romani populations by the effect of genetic drift alone. Rather, the high frequencies of U3-haplotypes observed in the Polish, Lithuanian and Spanish Roma allow us to suggest that members of a single Roma group migrated independently to the north and southwest of Europe. This scenario is also supported by Y-chromosome data indicating that the Lithuanian and Spanish Roma are characterized by high frequencies (25 and 33%, respectively) of a specific J2f-lineage, defined by the M67 marker (Gresham et al. 2001). This lineage has not been shown to be of European populations, but is present in populations from Pakistan, central Asia and the Middle East (Underhill et al. 2000; Kivisild et al. 2003). Taking into consideration the pattern of the geographic distribution of mtDNA and Y-chromosome haplotypes, it can be seen that mitochondrial haplogroup M5 and Y-chromosomal haplogroup H1 (defined by M82 marker) represent the genetic composition of the ancestral Roma population. Meanwhile, some DNA haplogroups are more restricted geographically, while some haplotypes correspond to the founding lineages of individual populations (subisolates) within the Roma groups. Thus, further genetic studies will be very useful to examine the population history of the Roma, as well as to reveal individual genetic subisolates suitable for the fine mapping of genes involved in complex disorders. Malyarchuk, B. A., et al. " Mitochondrial DNA diversity in the Polish Roma." Annals of human genetics 70.2 (2006): 195-206. |
|
|
|
Post by Admin on Nov 4, 2014 14:17:55 GMT
Only a few genetic studies have been carried out to date in Bolivia. However, some of the most important (pre)historical enclaves of South America were located in these territories. Thus, the (sub)-Andean region of Bolivia was part of the Inca Empire, the largest state in Pre-Columbian America. We have genotyped the first hypervariable region (HVS-I) of 720 samples representing the main regions in Bolivia, and these data have been analyzed in the context of other pan-American samples (>19,000 HVS-I mtDNAs). Entire mtDNA genome sequencing was also undertaken on selected Native American lineages. Additionally, a panel of 46 Ancestry Informative Markers (AIMs) was genotyped in a sub-set of samples. The vast majority of the Bolivian mtDNAs (98.4%) were found to belong to the main Native American haplogroups (A: 14.3%, B: 52.6%, C: 21.9%, D: 9.6%), with little indication of sub-Saharan and/or European lineages; however, marked patterns of haplogroup frequencies between main regions exist (e.g. haplogroup B: Andean [71%], Sub-Andean [61%], Llanos [32%]). Analysis of entire genomes unraveled the phylogenetic characteristics of three Native haplogroups: the pan-American haplogroup B2b (originated ~21.4 thousand years ago [kya]), A2ah (~5.2 kya), and B2o (~2.6 kya). The data suggest that B2b could have arisen in North California (an origin even in the north most region of the American continent cannot be disregarded), moved southward following the Pacific coastline and crossed Meso-America. Then, it most likely spread into South America following two routes: the Pacific path towards Peru and Bolivia (arriving here at about ~15.2 kya), and the Amazonian route of Venezuela and Brazil southwards. In contrast to the mtDNA, Ancestry Informative Markers (AIMs) reveal a higher (although geographically variable) European introgression in Bolivians (25%). Bolivia shows a decreasing autosomal molecular diversity pattern along the longitudinal axis, from the Altiplano to the lowlands. Both autosomes and mtDNA revealed a low impact (1–2%) of a sub-Saharan component in Bolivians. 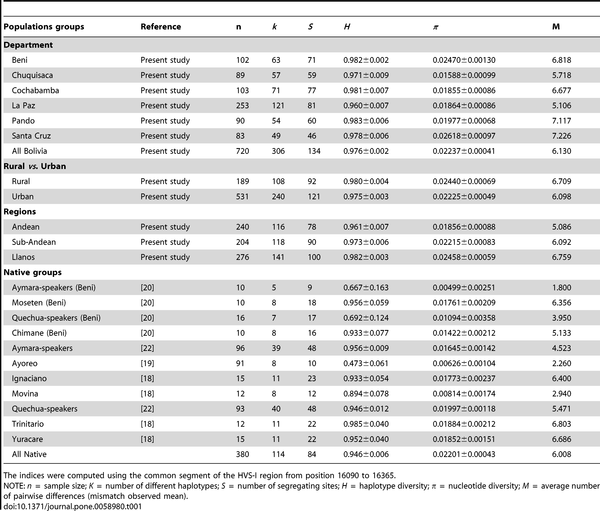 TABLE 1: Diversity indices in Bolivian mtDNAs and main American and African TABLE 1: Diversity indices in Bolivian mtDNAs and main American and AfricanSeveral diversity indices have been computed considering different hierarchical levels (Table 1). When analyzed by main regions, all seemed to show similar diversity values; with the Department of La Paz harboring the lowest haplotype diversity, and Chuquisaca being the region with the lowest nucleotide diversity. The highest haplotype diversity was found in the North (Pando) but the highest nucleotide diversity is observed in the South (Santa Cruz). Therefore, there is no obvious correlation between departments and molecular diversity as measured by way of statistical summary indices. When examining the diversity by rural and urban populations, it was observed that rural populations harbor higher nucleotide and haplotype diversity than urban populations. The most apparent geographic pattern was found when examining molecular diversity values by main ecological region. The Andean followed by the Sub-Andean regions had substantial lower diversity values than the Llanos. Therefore, the diversity was found to increase longitudinally, from the high mountains to the lowlands of the Llanos. Diversity was particularly low in some ethnic groups compared to general urban and rural populations. For instance, the Ayoreo (from Bolivia and Paraguay) and the Aymara had extremely low diversity values compared to the average values observed in Native Bolivians and Bolivians in general (Table 1), a fact that is most likely due to strong founder effect in the case of the Ayoreo [19], while in the Aymara discussed by Corella et al. [20] it was most likely due to the small sample size analyzed (the values were compared with those obtained with the Aymara population from Gayá-Vidal et al. [22] using a larger sample size). Our sample from general rural and urban Beni showed significantly higher diversity values than those observed from other Native American groups from the same department (e.g. the Piedmont populations of Moseten, Chimane, etc [20], the Llanos populations analyzed in [18]; see Table 1). 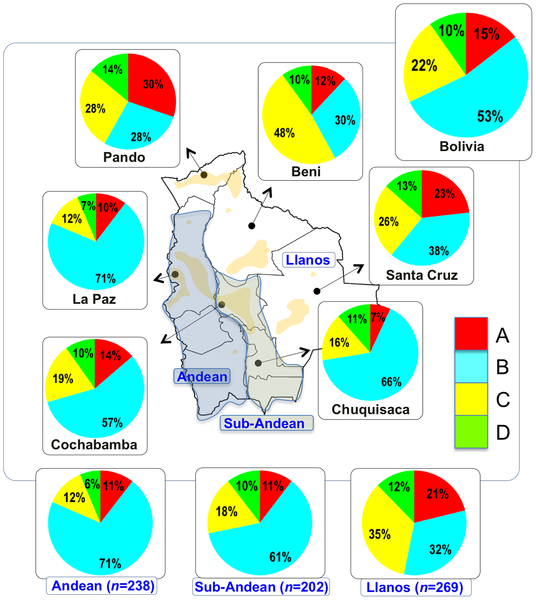 Figure 1. Map of Bolivia showing the location of the samples collected in the present study. Figure 1. Map of Bolivia showing the location of the samples collected in the present study.
The pie charts represent the distribution of basal Native American haplogroup frequencies in the region.The mtDNA Native American component predominates in the Bolivian population (98.4%); most of the variation can be classified into one of main Native American haplogroups, (A: 14.3%, B: 52.6%, C: 21.9%, D: 9.6%), Figure 1. There were remarkable differences in haplogroup frequencies between the main Bolivian departments (Figure 1). For instance, the department of La Paz had the following haplogroup composition, A: 10%, B: 71%, C: 12%, and D: 7%, which was in sharp contrast with the composition observed in the department of e.g. Beni, A: 12%, B: 30%, C: 48%, and D: 10%. The haplogroup distribution found by Afonso-Costa et al. [21] in a smaller sample from La Paz was slightly different to that found in the Bolivians from La Paz studied here, but both samples agreed regarding the high proportion of haplogroup B in this area. This geographic pattern mirrors in reality the location of the departments at different altitudes; thus, the most important differences are observed between Andean and Sub-Andean populations versus Llanos. For instance, haplogroup B is the predominant haplogroup in the Altiplano (71%), which decreases to 61% in Sub-Andean populations and 32% in Llanos (Figure 1). As expected, Bolivia shares more haplotypes with South America than with Meso and North America (Table 2; Table S2), although these values have to be interpreted with care due to the different sample sizes. Note however that the largest sample size is in North America, but Bolivia only shares 18% of the haplotypes with this region but 49% with other South American populations (Table 2). It is also worth mentioning that 48 haplotypes are shared between the three American regions; therefore, of the 68 haplotypes shared with North America, most of them are also shared with South America (Table 2, Table S2). A large amount of haplotypes has been only observed in Bolivia (250 out of 383 different haplotypes; 65%). Entire genomes from Native American BoliviansIn order to investigate the Native American lineages in our Bolivian samples further, we selected those showing a distinctive pattern from the control region data. Thus, we chose two mtDNAs carrying the tandem variants T16097C A16098G on top of the sequence motif for haplogroup A2, and a group of six mtDNAs all carrying transition G16145A on top of the B2 basal motif (Table S3). Entire genome sequencing revealed some interesting phylogeographic features of these mtDNAs. 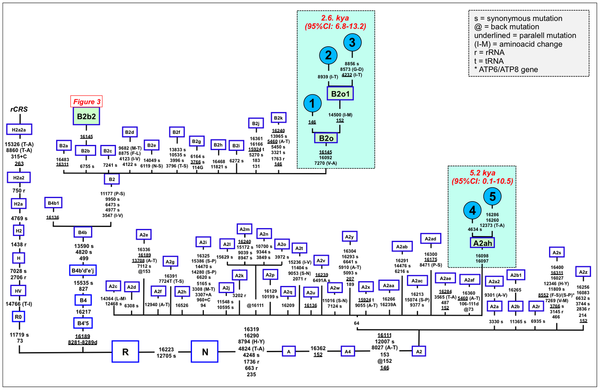 Figure 2. Maximum parsimony tree of the main branches characterizing haplogroups A2 and B2, indicating the new branches generated in the present study, namely, A2ah and B2o. Figure 2. Maximum parsimony tree of the main branches characterizing haplogroups A2 and B2, indicating the new branches generated in the present study, namely, A2ah and B2o.The tandem transitions T16097C A16098G conform per se a very distinctive motif that is found very rarely in mtDNA databases. Within haplogroup A2, this motif alone (without any other control or coding region variant) defines a novel branch of the Native American phylogeny, here named as A2ah (Figure 2). The two genomes show variability within this clade and, given its very restrictive location within Bolivia, this most likely point to an origin in Bolivia or the surrounding territories. We further investigated A2ah in public databases of entire mtDNA genomes. We found just one entire genome (JQ702082) that carries the transition A16098G on top of the A2 motif but it differs substantially from the two genomes found in Bolivia (apart from lacking the transition T16097C); therefore, its inclusion within A2ah should be considered to be very dubious. According to the entire genome sequences available, it can be said that haplogroup A2ah originated about 5,200 thousand years ago (kya) (although with a large 95%CI: 0.1–10.5). We additionally investigated the phylogeographic characteristics of these lineages by looking at the abundant amount of data available in the literature on control region segments. We only detected nine mtDNAs matching the motif of A2ah. Four out of ten were observed in the data reported by Behar et al. [61] but geographic information was not available. Two other HVS-I mtDNAs were found in the ‘Hispanic’ North American subset of the SWGDAM database [62]; three other A2ah mtDNAs were observed in South America, two among the Brazilian samples of Alves-Silva et al. [13], and one in the Toba from Gran Chaco (North Argentina) [63]. In Bolivia, only four A2ah members were found (0.5%), all of which were observed in the Llanos (three in Santa Cruz and one in the Beni department). In general, the HVS-I data suggest that this lineage could have originated in Bolivia or some place in Central South America. The two members observed in the ‘Hispanic’ samples from the SWGDAM could perfectly represent recent immigrations into USA. The motif T16097C A16098G was also observed in Mainland Scotland (Western Islands; Isla of Skye) and two other samples in [61], but none of the mtDNAs carried the variation defining haplogroup A2. A phylogenetic conflict exists when trying to reconstruct the most parsimonious tree of the six B2 mtDNAs carrying transition G16145A. Three of the genomes (#6, #7 and #8 in Figure 3) carried the synonymous transition G6755A, while three other genomes (#1, #2 and #3) lacked G6755A but carried the non-synonymous transitions T7270C and T16092C instead (Figure 2). Given that G16145A seems to have a much higher mutation rate than G6755A (22 versus 2 mutational hits in Soares et al. [41], and 24 versus 5 in Phylotree; respectively), we decided to resolved the phylogeny as shown in Figure 2 and Figure 3. 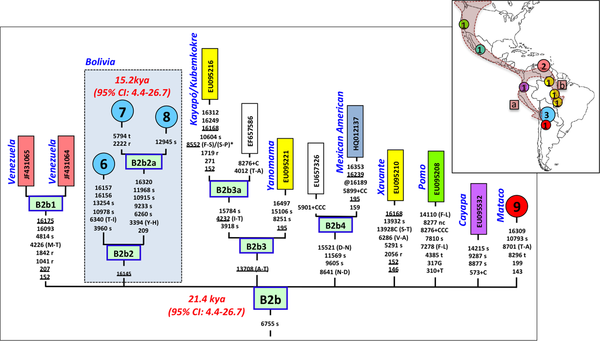 Figure 3. Maximum parsimony tree of haplogroup B2b. Figure 3. Maximum parsimony tree of haplogroup B2b.Thus, the motif T7270C T16092C G16145A defines a new branch of the Native American phylogeny named here as B2o, which is represented by three Bolivian genomes (Figure 2). No other entire genome was observed in public resources belonging to B2o. Dating the phylogeny of B2o (Figure 2) indicates that this haplogroup originated about 2.6 kya (95%CI: 6.8–13.2). Searching control region databases, we observed only two HVS-I candidate B2o sequences: one was observed in San Martin de Pangoa in Perú, a small town on the eastern slope of the Andes inhabited by Quechua and Nmatsiguenga people [64], while the other B2o sequence was observed in Guam (an island located in the western Pacific Ocean, territory of USA). The latter, however, carries the motif T16189C T16217C A16247G C16261T, which is very common in the Micronesia, Australia, etc. [65], [66] and points to a different clade of the worldwide phylogeny, haplogroup B4a. In Bolivia, we found seven B2o representatives (1%), most of them in the Llanos (five in Pando and one in Santa Cruz departments), and one in a Sub-Andean locality of La Paz. Therefore, altogether the data suggest that this lineage has also originated in Bolivia or some nearby region, most likely located in the Andes. The entire genomes of the three Bolivian mtDNAs carrying G16145A but lacking T16092C, all share the transition G6755A. By searching the databases on entire genomes for the transition G6755A, we observed 10 additional mtDNAs belonging to this branch. In reality, this clade had already been described in the recent literature [6], [7] and was named B2b. However, its internal variation was never analyzed in detail. Figure 3 shows the phylogeny of B2b and the geographical location where the entire genomes were observed. Geographic and/or ethnic affiliation was available for eleven entire genomes (including the Bolivian ones): one Pomo (North California, USA) [8], one Mexican American [67], two Venezuelans from Pueblo Llano [68], one Yanomama (Brazil, Venezuela) [8], one Cayapa (Equador) [6], one Kayapó/Kubemkokre (South Amazonia, Brazil) [8], one Xavante (Brazil) [8], and three Bolivians (present study). We additionally detected one entire genome in our DNA databank in a Mataco Native from North Argentina, which was added to the phylogeny of Figure 3 (#9). According to the phylogeny of B2b in Figure 3, B2b appeared about 21.4 kya (95% CI: 4.4–26.7). The Bolivian sub-clade B2b2 is much younger, approximately 15.2 kya (95%CI 4.4–26.7). In the large HVS-I database, we only observed 14 B2b candidates, almost overlapping the territories represented by the entire genomes. In Bolivia, we counted seven B2b candidates: one Andean (La Paz), four Sub-Andean (Cochabamba, La Paz and Chuquisaca), and two in the Llanos (Beni and Santa Cruz). Continental ancestry in BoliviansA panel of 46 AIMS was genotyped in Bolivians in order to infer their main continental ancestry. Data from main continental reference samples (Africa, East Asia, Europe and America) were used as classification sets. 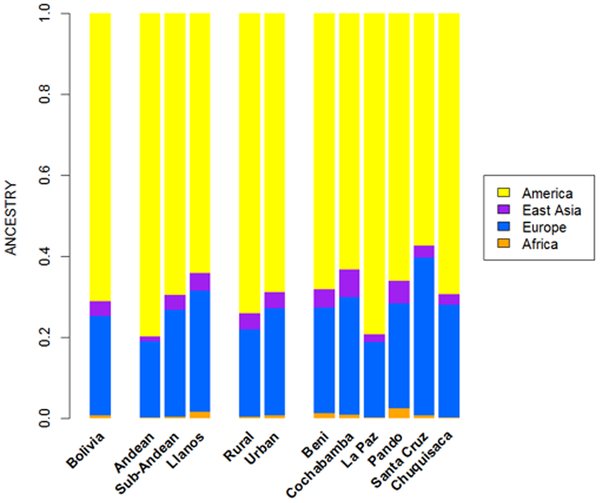 Figure 4. Average continental ancestry of Bolivians analyzed using a panel of 46 AIMs. These values are obtained from the STRUCTURE analysis and the optimal value K = 4. Figure 4. Average continental ancestry of Bolivians analyzed using a panel of 46 AIMs. These values are obtained from the STRUCTURE analysis and the optimal value K = 4.Analysis of ancestry was carried out using STRUCTURE. Figure 4 summarizes the average continental ancestries observed in Bolivians under different grouping schemes, while Figure 5 shows the STRUCTURE bar-plots. The analysis showed that, on average, 71% of the component in the total Bolivian sample is Native American, followed by 25% of European ancestry. When examining the ancestry by departments, La Paz was the region that showed the highest Native American ancestry (79%) (Figure 4 and Figure 5), and, therefore, the lowest European component (19%). On the other side is the department of Santa Cruz, with 57% of Native American ancestry and 39% European. The African component was very low in all of the departments, showing the highest value in the department of Pando (2.5%), in the North. When examining by ecological regions, it was also evident that the Native American component is higher in the mountainous West (80%), and decreases progressively eastward: sub-Andean (70%) and Llanos (64%). The differences are less apparent when examining rural vs. urban areas (74% and 69% of Native American component, respectively). 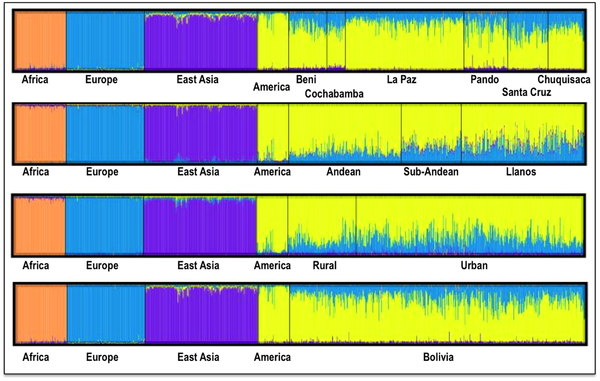 Figure 5. STRUCTURE analysis of Bolivians based on the 46 AIMs panel genotyped in the present study. Figure 5. STRUCTURE analysis of Bolivians based on the 46 AIMs panel genotyped in the present study.The STRUCTURE bar-plot of Figure 5 indicates the ancestral membership for each individual in the source population and the Bolivian samples. It clearly shows that most of the individuals have a Native American ancestry, with only a few exceptions. The high European component of Santa Cruz compared to other departments is also evident in this bar-plot, and is also evident in the Andeans. When looking at the most extreme values, we found that there are only a few that have European ancestry above 50% in most of the departments. It is, however, rare to see individuals with high African membership; the highest values correspond to 23% in one individual from La Paz and another one from Pando (17%). The membership of individuals into the East Asian cluster is significantly higher in some individuals, as is also evident in the STRUCTURE plots. However, this perhaps mirrors the fact that the separation between the Native American component and the East Asian one is not perfect. The overlap between Native Americans and East Asians can be observed more clearly in the PCA analysis (see below). This fact has been already observed in Pereira et al. [37]. Thus, most of the East Asian component would be captured by the Native American cluster in cases using a three-group structure analysis (Native American, African, and Europe). 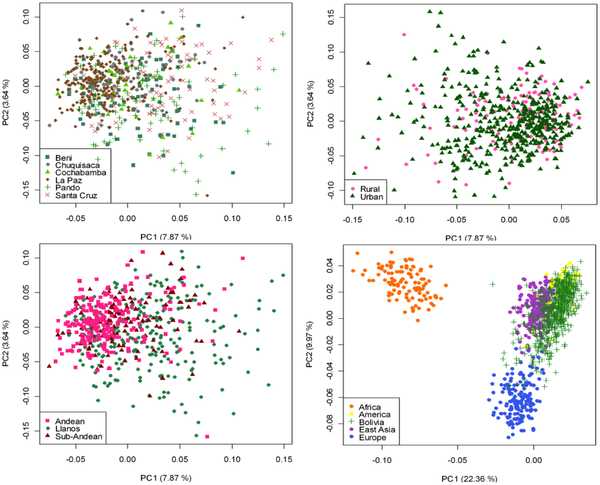 Figure 6. PCA of Bolivian profiles based on the 46-AIMs panel genotyped in the present study. Each figure shows different grouping schemes mainly aiming to show the within-Bolivian diversity. Figure 6. PCA of Bolivian profiles based on the 46-AIMs panel genotyped in the present study. Each figure shows different grouping schemes mainly aiming to show the within-Bolivian diversity.The PCA agrees well with the results observed in STRUCTURE. The Bolivian profiles group mainly with the Native American ones from HapMap, with only some samples showing a projection towards the European cluster (Figure 6). The minor African component detected in the STRUCTURE analysis seems not to be relevant in the PCA; only one sample show some affinity to the African cluster; it is in fact the same sample with a 23% African ancestry in STRUCTURE. The PCA carried out by departments (Text S1) showed that the department of La Paz (Andes) was more tightly grouped than the profiles from other departments; in the other pole are the departments of Santa Cruz and Pando, showing more dispersed patterns (Llanos). This west-east pattern is more evident when observing the PCA by main regions (Figure 6): the Andean profiles are more tightly grouped in one pole of the plot, while the profiles from Llanos are in the other pole and are more dispersed, with the sub-Andean profiles occupying an intermediate position between East and West. This dispersion is also mirrored when examining the standard deviation (SD) of the Native American membership values in the main regions: (i) Andean: mean = 79.7%, SD = 0.086%; (II) sub-Andean: mean = 69.6%, SD = 0.09; and (iii) Llanos: mean 64.2%, SD = 0.137% (Table S4); given that the department of La Paz is basically an Andean department, it also shows the lowest SD (0.85%). Discussion
Bolivia shows a mainly Native American mtDNA component (98%). Only 1.5% of the profiles have Sub-Saharan mtDNA ancestry. The impact of Europeans in the Bolivian mtDNA pool is minimal (0.5%), which also contrasts with other South American locations [11], [27]. Although the Native American mtDNA component predominates in the country, there is a highly diverse and geographic stratification in the country. In a large geographical scale, the largest difference corresponds to Llanos vs. the Andean and Sub-Andean regions. The political definition of the departments overlap quite well with the main latitudes observed in the country, which would explain the correspondence observed when carrying out different statistical analysis. Molecular diversity is extremely low in some indigenous group compared to urban and other rural populations, suggesting the existence of important episodes of isolation and genetic drift in Native communities. AMOVA carried out at Bolivian populations to different hierarchical levels allowed a better understanding of the spatial geographic patterns of variability in large regions. The results agree that most of the variation accounts within populations, but that the major differentiation occurs between the Andes and the Llanos. In addition, some mtDNA phylogeographic features indicate the presence of common lineages between different Andean regions in Peru and Bolivia, most likely testifying for the common demographic past during the Inca’s Empire period. This continuity in the Andean region was also observed in the Aymaras and Quechuas analyzed by Gayá-Vidal et al. [22]. By way of sequencing the entire genome of nine mtDNAs (eight from Bolivia and one from a northern Argentinean Native Mataco), and compiling a large amount of data from the literature, we could shed light on three Native American lineages: A2ah, B2o and B2b. The three haplogroups are rare in Bolivia (ranging from 0.5 and 1%) and are very rare continentally. The three clades show significant diversity within the Bolivian country, indicating that they most likely evolved locally after their arrival from other neighboring regions. The clade for which there are more data available (literature and present study) is B2b. This haplogroup most likely arose in North California; however, as suggested by its TMCRA (~21.4 kya; see also [67]) and its phylogeographic characteristics, it could have been originated even in the north most region of the American continent, constituting a new minor Paleo-Indian founder. The data also indicate that B2b traveled South, most likely following the Pacific coastline (the main route followed by the First Americans) [4], [5], [9], [69]. It probably entered the South American sub-continent following two different paths: (i) travelling further south following the Pacific side, then reaching Equador, Peru, and Bolivia; and (ii) following firstly an eastwards direction, and secondly southwards crossing the Amazon basin in Venezuela and then Brazil (or firstly bordering the Atlantic coast southwards). At the very least, these lineages arrived in northern Argentina, but it is likely that further sampling will probably detect B2b in the most southern edge of the continent. The data indicate that the Bolivian sub-lineage B2b2 arrived in this region about 15,000 years ago. Ancestry analyses carried out on a panel of AIMs indicated that Bolivians have a substantial European ancestry (although a possibility exists that this proportion could be over-estimated; see above), which varies substantially between departments, thus indicating a differential regional impact of the European Colonialism in Bolivia. The African component is very low (in agreement with the mtDNA) as compared to other South American populations, such as the Caribbean coast [17], [70], Colombia [16], and Brazil [13], but is comparable to others such as Argentina [10], [11], [27]. It must be taken into account that the present study did not include samples from the Yungas, a small province located within the department of La Paz that seems to concentrate the main African component in Bolivians. The African component of the Yungas people is not only inferred from their distinguishable African cultural and biological features, but also from the genetic inferences made using panels of AIMs analyzed in a small group of people from this region [23]. The present study represents the largest and most comprehensive analysis of genetic variation investigated to date in rural and urban Bolivians from the point of view of mtDNA and autosomal data. The results indicated that even those Bolivians that did not self-identify as belonging to a particular Native America ethnic group still preserved a main Native American genetic character in their genomes. Deep analyses of selected mtDNA genomes also indicate the presence of lineages that appears to be autochthonous to these peoples; provided that these lineages have not been identified in large American databases. The Native American component of Bolivians is significantly higher when observing the mtDNA instead of the autosomes. This is a common feature in other South and Central American regions (e.g. Argentina [10], Panama [2])where the maternal Native American component has been found to be much better preserved in the maternal specific genome, a fact that is generally explained by the higher proportion of European males arriving to America than females. Taboada-Echalar P, Álvarez-Iglesias V, Heinz T, Vidal-Bralo L, Gómez-Carballa A, et al. (2013) The Genetic Legacy of the Pre-Colonial Period in Contemporary Bolivians. PLoS ONE 8(3): e58980. doi:10.1371/journal.pone.0058980 |
|
|
|
Post by Admin on Nov 22, 2014 15:09:58 GMT
 Recent studies of Y-chromosomes and mtDNA of Icelanders indicate that this population's gene pool was the result of admixture between male subjects whose patrilineal ancestry lay in Scandinavia, and female subjects whose matrilineal ancestry lay within the British Isles (Helgason et al, 2000a, 2000b, 2001). A further study suggests that Icelanders and Orkney Islanders have similar proportions of Scandinavian mtDNA ancestry (approximately36%; Helgason et al, 2001). Other studies of the genetic history of the North Atlantic region have concentrated on discerning the different proportions of northwestern European, Anglo-Saxon and Scandinavian patrilineal ancestry using Y-chromosome data (Wilson et al, 2001; Weale et al, 2002; Capelli et al, 2003). A sizeable component of Scandinavian patrilineal ancestry has been reported in Orkney (55%) and Shetland (68%) based on likelihood estimates of population admixture and principal components analyses of haplotype frequencies (Capelli et al, 2003). Here, we present an analysis of new mtDNA and Y-chromosome data from Shetland and new Y-chromosome data from Scotland, Norway, the Western Isles and the Isle of Skye. We combine our new data with those that have been previously published in order to investigate further the relationships among populations of the North Atlantic, and to establish what genetic legacy remains from Scandinavian activities in this area during the Viking period. In particular, our aim is to evaluate the different genetic impact of Scandinavian male and female subjects on Shetland and other island and coastal populations in the North Atlantic region.  Scandinavian and British/Irish ancestry in the island and coastal populations of the North Atlantic region Scandinavian and British/Irish ancestry in the island and coastal populations of the North Atlantic regionThe analysis of population structure presented above indicates significant differences between the Y-chromosome and mtDNA pools of the two putative parental populations of Britain/Ireland and Scandinavia (represented by the contemporary inhabitants of Norway, Sweden and Denmark, and Scotland and Ireland, respectively). As a consequence, it should be possible to distinguish between the contributions that each of the two parental populations has made to the Y-chromosome and mtDNA gene pools of the island and coastal populations of the North Atlantic region. Estimates of population admixture can be made using one of several different approaches; as a consequence, the choice of statistical method used here necessitates some comment. Standard frequency-based methods of admixture estimation assume that all alleles present in the hybrid population are also present in at least one of the putative parental populations (Roberts and Hiorns, 1965; Long, 1991). Such methods are unlikely to be informative for the current molecular data sets because as many as half of the Y-chromosome and mtDNA haplotypes in the hybrid populations are not found in either of the parental populations. An alternative method, which is based on average mutational differences between haplotypes drawn within or between admixed and parental populations, does not require any of the haplotypes from the hybrid population to be present in the parental populations. However, this method is only effective in cases where there is a marked mutational divergence between haplotypes from the parental populations (Bertorelle and Excoffier, 1998). This approach is not appropriate for the current study because the phylogenetic divergence of haplotypes from the parental populations is negligible, particularly for mtDNA.  Figure 1. Map showing the proportions of Scandinavian and British/Irish ancestry for mtDNA (Mt) and Y-chromosomes (Y) for each of the admixed populations from the North Atlantic region included in this study. The Scottish island groups of Shetland, Orkney, the Western Isles and Skye, and the region that we define as the 'North and West coast of Scotland', are encircled for clarity. Figure 1. Map showing the proportions of Scandinavian and British/Irish ancestry for mtDNA (Mt) and Y-chromosomes (Y) for each of the admixed populations from the North Atlantic region included in this study. The Scottish island groups of Shetland, Orkney, the Western Isles and Skye, and the region that we define as the 'North and West coast of Scotland', are encircled for clarity.An alternative method, mrho, was used in order to circumvent the problems inherent in the methods described above. mrho is a heuristic, frequency-based admixture approach that uses a distance matrix between haplotypes to identify founder haplotypes in the parental populations for all private haplotypes in hybrid populations. This method also takes into account uncertainty in estimates of haplotype frequencies in the hybrid and parental populations due to either sampling variance or genetic drift subsequent to population mixing (Helgason et al, 2001). Table 3 and Figure 1 show estimates of Scandinavian and British/Irish ancestry based on the mrho approach. The proportion of Scandinavian patrilineal (Y-chromosome) ancestry is highest in Iceland (75%), is somewhat lower in Shetland (44.5%) and Orkney (31%), declines further in the Western Isles and Skye (22.5%) and is lowest along the N&W coastline of Scotland (15%). This is consistent with a lasting genetic legacy of Scandinavian males in both Orkney and Shetland with a lesser, but still measurable, contribution to the other Scottish locations. The proportions of Scandinavian matrilineal (mtDNA) ancestry estimated for Shetland, Orkney and the N&W coastline of Scotland are almost identical to their patrilineal counterparts. In contrast, the estimated contribution of Scandinavian female subjects to the Western Isles and Isle of Skye (11%) is half that of Scandinavian male subjects (approx22%). This discrepancy is even more marked for Icelanders, whose effective founding population appears to have consisted largely of female subjects with a British/Irish ancestry and male subjects with a Scandinavian ancestry (Helgason et al, 2000a, 2001). Our estimates of Scandinavian patrilineal ancestry for Shetland, Orkney and the Western Isles are smaller than those proposed by Capelli et al (2003), which were based on a different data set (their estimates were 68.3, 55.3 and 61.6%, respectively). We note, however, that while they employed a frequency-based admixture method that accounted for sampling variance and the impact of genetic drift, this method ignored all private haplotypes in the hybrid populations. It is likely that the difference between our admixture results and those of Capelli et al (2003) stems in part from this omission of private haplotypes. However, the difference could to some extent also be due to the use by Capelli et al (2003) of Y-chromosomes from both central Ireland and the Basque region to represent the 'Celtic' parental population.  Discussion DiscussionOur mitochondrial and Y-chromosome data suggest that Shetland has almost identical proportions of Scandinavian matrilineal and patrilineal ancestry (approx44%). The same balance is to be found in Orkney and the N&W coastline of Scotland, although the average Scandinavian contribution in these two areas is lower (approx30% in Orkney and approx15% in the N&W Scottish coastline). The results strongly suggest that the Scandinavian genetic contribution (in terms of both matrilineal and patrilineal ancestry) is greatest in those areas that lie close to Scandinavia. While it is possible that genetic drift has affected gene frequencies so that our estimates of admixture no longer accurately reflect the composition of founder populations, it seems unlikely to be coincidental that the overall proportion of Scandinavian ancestry for these three locations decreases in harmony with geographic distance from Scandinavia. Admixture analysis (using mrho) of recently published Y-chromosome data from the Faroe Islands (Jorgensen et al, 2004) also provides evidence for a trend of increased Scandinavian patrilineal ancestry in regions further from the British Isles. The estimated Scandinavian genetic component of the Faroese samples is 87% (N=89, k=26), a higher figure than for either Shetland or Orkney. Furthermore, as expected for a region with a historical population size even smaller than that of the Icelanders and with a similar degree of geographical isolation, our estimates suggest that the Faroese have the least diverse Y-chromosome pool (gene diversity=0.9298, thetak=11.99). Historical, archaeological, place-name and linguistic evidence indicates complete Norse cultural dominance of Shetland and Orkney during the Viking period (Jones, 1984). Although more distant from the Scandinavian power-base, Orkney was the nucleus of Scandinavian political and cultural influence in the North Atlantic colonies (Davies, 1999). This implies a Scandinavian presence in Orkney that was at least as great as that in Shetland. If this is the case and the indigenous pre-Scandinavian populations of Shetland and Orkney were about the same size, then the lesser Scandinavian genetic component in Orkney may be explained by greater post-Scandinavian migration from Scotland to Orkney than to Shetland. However, a smaller indigenous pre-Scandinavian population in Shetland, followed by similar numbers of Scandinavian colonizers and post-Scandinavian Scottish settlers would also account for the difference between the two islands. The genetic evidence does not allow us to determine whether the observed geographic gradient of Scandinavian ancestry is largely due to either variable pre-Scandinavian population sizes, variable numbers of Scandinavian settlers, variable levels of post-Scandinavian Scottish resettlement or some combination of all of these factors. However, the observed symmetry between Scandinavian mtDNA and Y-chromosome ancestry in Shetland, Orkney and the N&W coastline of Scotland suggests that roughly equal numbers of Scandinavian male and female subjects were involved in the colonization of these locations. In other words, the genetic evidence indicates a family-based Scandinavian settlement of Shetland, Orkney and the N&W coastline of Scotland. This contrasts with results obtained for the Western Isles/Isle of Skye and Iceland where estimated male and female Scandinavian components differ. The Western Isles/Isle of Skye have the lowest estimated proportion of Scandinavian ancestry (17%) while Iceland has the highest (55%), but both exhibit a two-fold excess of Scandinavian patrilineal ancestry relative to Scandinavian matrilineal ancestry. The data seem to suggest that the settlement pattern of both the Western Isles/Isle of Skye and Iceland included, in addition to Scandinavian family groups, a substantial number of males who took wives from indigenous British populations. Islands off the coast of Britain are relatively distant from the Scandinavian homeland and must have been viewed as frontier areas within the North Atlantic region during the Viking period. Iceland, an uninhabited island at the margin of the then known world, was even more remote. Linguistic, archaeological and historical evidence indicates that Scandinavian dominance was never complete or secure in places such as the British Isles (Davies, 1999). Viewed in this light, the excess of Scandinavian patrilineal ancestry observed in the Western Isles and Iceland and the symmetric contribution of Scandinavian male and female subjects in Shetland, Orkney and the N&W coastline of Scotland are consistent with a common feature of human colonizing behavior: This is that migration to insecure frontier areas tends to involve a disproportionate number of lone male colonizers, whereas family groups are more likely to be abundant in secure areas that are closer to the strongholds of colonial power. Goodacre, S., et al. " Genetic evidence for a family-based Scandinavian settlement of Shetland and Orkney during the Viking periods." Heredity 95.2 (2005): 129-135. |
|

