|
|
Post by Admin on Jun 1, 2022 17:56:04 GMT
 Figure 3 - figure supplement 1. Population structure of Italy during the Imperial Roman and Late Antiquity period. Ancient Italian genomes (colored points) from the Imperial Roman and Late Antiquity period were projected onto principal components of present-day genomes (gray points, populations labeled in Figure 2 - figure supplement 1). Present-day Italian genomes are highlighted by a gray filled ellipse. Star points are outliers and circle points are non-outliers. Outlier clusters that can be modeled using contemporaneous populations are labeled with the potential source region.  Figure 5 - figure supplement 1. Lack of sex-bias amongst outliers with valid qpAdm sources. The proportions of males and females do not differ significantly between outlier and non-outlier groups (p = 0.4256). The fisher exact test also produces an insignificant p-value.  Figure 5 - figure supplement 2. Distances of outliers to their candidate sources. Geographic distance between the sampling locations of “outlier with source” and the location of their putative source was estimated for each outlier. The mean distance was calculated if there were multiple putative sources. |
|
|
|
Post by Admin on Jun 1, 2022 21:58:30 GMT
 Figure 5 - figure supplement 3. Example routes and travel times across the Roman Empire. Routes and travel times were approximated using orbis.stanford.edu, a geospatial network model of the Roman Empire. Routes shown are the fastest routes during Summer for civilians, utilizing road, river, coastal sea, and open sea, and by foot if on road. Routes for military individuals (not shown) are marginally faster.  Figure 7 - figure supplement 1. A sigmaDisp - N parameter pair was chosen to closely approximate the observed FSTmax of ∼0.03 using grid search across a range of parameter pairs. We used the pair N = 50,000 & sigmaDisp = 0.02 for all other simulations we report.  Figure 7 - figure supplement 2. A sigmaDispLR parameter was chosen to qualitatively resemble long-range dispersal distances observed in the data, by comparing the distribution of distances under long-range dispersal (outliers) to randomly chosen distances given the spatial distribution of samples. We used a value of 0.20 for all other simulations we report. |
|
|
|
Post by Admin on Aug 19, 2022 1:28:39 GMT
In a series of articles published in the journal Nature Genetics, researchers have used data from the SPARK (Simons Powering Autism Research) research cohort, which was created to advance our understanding of the complex genetics of autism and includes genetic data from nearly 43,000 people with autism.
The findings show differences in genetic influences among people all along the autism spectrum.
“Autism is a spectrum, and includes individuals with profound autism who often have cognitive differences and/or epilepsy, as well as individuals who are talented and exceptional, often in specific areas.
“We are now appreciating that the genetic contributions to different phenotypes vary in terms of the genes involved; when those genes are activated during brain development; and how common some of the genetic variants are in the population,” said Wendy Chung, M.D., Ph.D., principal investigator of SPARK.
One study, “Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate effect genes,” was published in Nature Genetics on August 18, 2022. Researchers analyzed the DNA of almost 43,000 people with autism, including 35,000 participants from the SPARK autism research study, as part of SPARK’s ongoing effort to understand the full spectrum of autism genetics.
This largest-ever autism cohort allowed researchers to identify a group of novel “moderate-effect” genes that tend to contribute to autism through inherited variants.
It is widely known that autism is heritable, but previous studies have primarily identified autism genes with de novo variants (DNV)—variants that occur spontaneously in germ cells prior to conception—that are not inherited. Most of these variants are also implicated in other neurodevelopmental disorders (NDDs).
Most genetic variants of this type associated with autism have profound effects on the brain in those individuals when they occur. However, only 20 percent of individuals with autism have this type of genetic variant.
“For many years, we have known from twin studies that there must be inherited genetic variants that lead to autism, but we have not been able to systematically identify individual genes until now,” said lead author Pamela Feliciano, Ph.D., SPARK’s scientific director.
“We have now identified a group of genes associated with autism, that can include inherited variants, which begin to explain a different part of the autism spectrum.”
To gain a better understanding of the full spectrum of autism genes, the researchers analyzed 19,843 participants with autism, along with one or both of their biological parents, and found that roughly 20 percent of people with autism have de novo genetic variants that affect the function of the associated gene.
Nearly 70 percent of this genetic contribution can be attributed to known autism or neurodevelopmental disorder genes. However, this means that although known autism-associated genes are responsible for the majority of de novo variants, there are others still to be identified.
The researchers next added in another 22,764 individuals with autism and 236,000 people without autism from the general population. In this meta-analysis, they identified 60 autism genes whose contribution to autism is largely driven by rare inherited loss of function (LOF) variants transmitted by parents who do not have cognitive differences or autism. Of these genes, five have not previously been implicated in neurodevelopmental conditions.
Individuals with autism who carry inherited variants in these “moderate effect” genes are less likely to have cognitive differences than people with autism who carry LOF variants in well-established autism genes, such as CHD8 and SCN2A.
“The majority of parents who passed down these genetic variants in our study do not have cognitive differences or autism, but we know that these genes are associated with autism because we find that these variants are more frequently inherited by children with autism.
“We hypothesized that people with autism who have these inherited genetic variants are not as likely to have seizures and cognitive differences as people with de novo genetic variants. So far our data strongly supports this hypothesis,” said Dr. Feliciano.
The SPARK cohort’s reach
A second study also published in Nature Genetics, “Rare coding variation provides insight into the genetic architecture and phenotypic context of autism,” led by a team of investigators supported by the Simons Foundation Autism Research Initiative (SFARI) and the Autism Sequencing Consortium (ASC), performed analyses on genetic data from 20,627 people with autism, with new genetic data derived primarily from SPARK.
The team developed new methods to discover gains and losses of DNA, or copy number variants (CNVs), from exome sequencing, and methods to integrate data from these CNVs with other classes of de novo and rare inherited variants, and identified 72 genes associated with autism. Most evidence came from de novo variants, with smaller but significant contributions from rare inherited variants.
The researchers then combined data from the autism studies with a large dataset of 31,000 families in which the child was diagnosed with developmental delay and/or other neurodevelopmental conditions.
These analyses discovered 373 genes associated with these diverse neurodevelopmental outcomes, and allowed the team to identify genes more associated with autism than with other neurodevelopmental conditions, and vice versa.
They found that genes associated predominantly with developmental delay tend to be important in early neuronal development, whereas autism genes tend to play a role in more mature neurons.
The study’s senior author, Michael Talkowski, Ph.D., director, Center for Genomic Medicine at Massachusetts General Hospital and member of the Broad Institute, noted that “the scale of the data collections from SPARK, the ASC and other sources—as well as the newly developed methods—has allowed us to explore the relative contribution of the diverse classes of genetic variants that contribute to a continuum of neurodevelopmental variability across these datasets.”
|
|
|
|
Post by Admin on Aug 19, 2022 6:06:02 GMT
Article Published: 18 August 2022 Rare coding variation provides insight into the genetic architecture and phenotypic context of autism Jack M. Fu, F. Kyle Satterstrom, …Michael E. Talkowski Nature Genetics (2022) 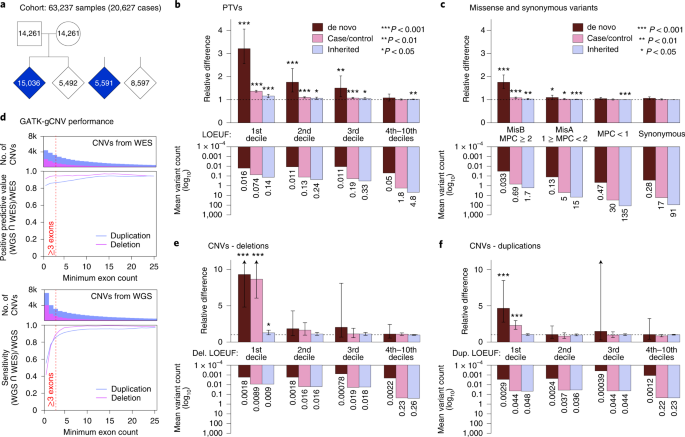 Abstract Some individuals with autism spectrum disorder (ASD) carry functional mutations rarely observed in the general population. We explored the genes disrupted by these variants from joint analysis of protein-truncating variants (PTVs), missense variants and copy number variants (CNVs) in a cohort of 63,237 individuals. We discovered 72 genes associated with ASD at false discovery rate (FDR) ≤ 0.001 (185 at FDR ≤ 0.05). De novo PTVs, damaging missense variants and CNVs represented 57.5%, 21.1% and 8.44% of association evidence, while CNVs conferred greatest relative risk. Meta-analysis with cohorts ascertained for developmental delay (DD) (n = 91,605) yielded 373 genes associated with ASD/DD at FDR ≤ 0.001 (664 at FDR ≤ 0.05), some of which differed in relative frequency of mutation between ASD and DD cohorts. The DD-associated genes were enriched in transcriptomes of progenitor and immature neuronal cells, whereas genes showing stronger evidence in ASD were more enriched in maturing neurons and overlapped with schizophrenia-associated genes, emphasizing that these neuropsychiatric disorders may share common pathways to risk. www.nature.com/articles/s41588-022-01104-0 |
|
|
|
Post by Admin on Dec 9, 2022 5:39:48 GMT
The researchers used ancient genomes to reveal new information about the human history of adaption.
Ancient DNA, including samples from human remains that are around 45,000 years old, has helped researchers understand a previously unknown aspect of humanity’s evolution.
The new research, which was co-led by Dr. Yassine Souilmi, Group Leader at the Australian Centre for Ancient DNA at the University of Adelaide, was recently published in the journal Nature Ecology and Evolution.
“It was widely believed the genetics of our human ancestors didn’t change due to environmental pressures as much as other animals, due to our enhanced communication skills and ability to make and use tools,” Dr. Souilmi said.
“However, by comparing modern genomes with ancient DNA, we discovered more than 50 cases of an initially rare beneficial genetic variant becoming prevalent across all members of ancient human groups. In contrast to many other species, evidence for this type of adaptive genetic change has been inconsistent in humans. This discovery consequently challenges the prevailing view of human adaptation, and gives us a new and exciting insight into how humans have adapted to the novel environmental pressures they encountered as we spread across the planet.”
Examining ancient DNA has been crucial in revealing the secrets of human evolution, according to co-lead author Dr. Ray Tobler, an Adjunct Fellow at the University of Adelaide and a DECRA fellow at the Australian National University.
“We believed historical mixing events between human groups might have hidden signs of genetic changes in modern human genomes,” Dr. Tobler said.
“We examined DNA from more than 1,000 ancient genomes, the oldest which was around 45,000 years old, to see if certain types of genetic adaptation had been more common in our history than studies of modern genomes had suggested.”
Professor Christian Huber, a senior author of the research paper, is an Adjunct Fellow at the University of Adelaide and an Assistant Professor at Penn State University.
“The use of ancient genomes was crucial because they preceded major historical mixing events that have radically reshaped modern European genetic ancestry,” Professor Huber said.
“This allowed the recovery of historical signs of adaptation that are invisible to standard analysis of modern genomes.”
Reference: “Admixture has obscured signals of historical hard sweeps in humans” by Yassine Souilmi, Raymond Tobler, Angad Johar, Matthew Williams, Shane T. Grey, Joshua Schmidt, João C. Teixeira, Adam Rohrlach, Jonathan Tuke, Olivia Johnson, Graham Gower, Chris Turney, Murray Cox, Alan Cooper, and Christian D. Huber, 31 October 2022, Nature Ecology & Evolution.
DOI: 10.1038/s41559-022-01914-9
|
|

