|
|
Post by Admin on Jan 15, 2023 4:18:23 GMT
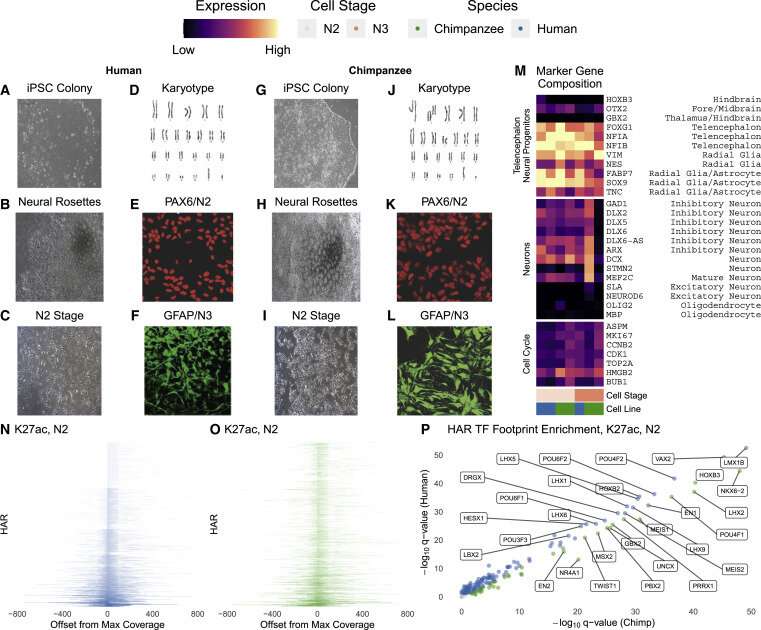 Humans and chimpanzees differ in only one percent of their DNA. Human accelerated regions (HARs) are parts of the genome with an unexpected amount of these differences. HARs were stable in mammals for millennia but quickly changed in early humans. Scientists have long wondered why these bits of DNA changed so much, and how the variations set humans apart from other primates. Now, researchers at Gladstone Institutes have analyzed thousands of human and chimpanzee HARs and discovered that many of the changes that accumulated during human evolution had opposing effects from each other. "This helps answer a longstanding question about why HARs evolved so quickly after being frozen for millions of years," says Katie Pollard, Ph.D., director of the Gladstone Institute of Data Science and Biotechnology and lead author of the new study published today in Neuron. "An initial variation in a HAR might have turned up its activity too much, and then it needed to be turned down." The findings, she says, have implications for understanding human evolution. In addition—because she and her team discovered that many HARs play roles in brain development—the study suggests that variations in human HARs could predispose people to psychiatric disease. "These results required cutting-edge machine learning tools to integrate dozens of novel datasets generated by our team, providing a new lens to examine the evolution of HAR variants," says Sean Whalen, Ph.D., first author of the study and senior staff research scientist in Pollard's lab. Enabled by machine learning Pollard discovered HARs in 2006 when comparing the human and chimpanzee genomes. While these stretches of DNA are nearly identical among all humans, they differ between humans and other mammals. Pollard's lab went on to show that the vast majority of HARs are not genes, but enhancers— regulatory regions of the genome that control the activity of genes. More recently, Pollard's group wanted to study how human HARs differ from chimpanzee HARs in their enhancer function. In the past, this would have required testing HARs one at a time in mice, using a system that stains tissues when a HAR is active. Instead, Whalen input hundreds of known human brain enhancers, and hundreds of other non-enhancer sequences, into a computer program so that it could identify patterns that predicted whether any given stretch of DNA was an enhancer. Then he used the model to predict that a third of HARs control brain development. "Basically, the computer was able to learn the signatures of brain enhancers," says Whalen. Knowing that each HAR has multiple differences between humans and chimpanzees, Pollard and her team questioned how individual variants in a HAR impacted its enhancer strength. For instance, if eight nucleotides of DNA differed between a chimpanzee and human HAR, did all eight have the same effect, either making the enhancer stronger or weaker? "We've wondered for a long time if all the variants in HARs were required for it to function differently in humans, or if some changes were just hitchhiking along for the ride with more important ones," says Pollard, who is also chief of the division of bioinformatics in the Department of Epidemiology and Biostatistics at UC San Francisco (UCSF), as well as a Chan Zuckerberg Biohub investigator. 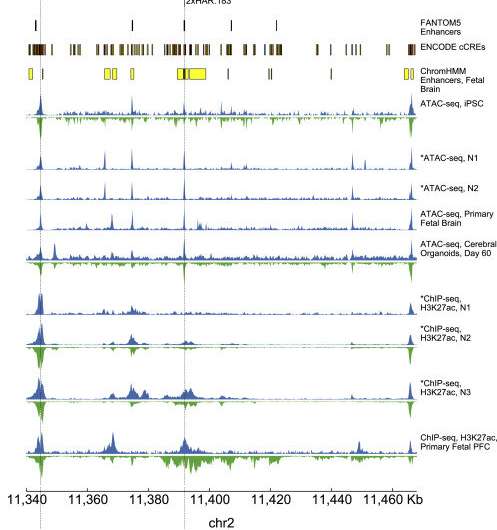 To test this, Whalen applied a second machine learning model, which was originally designed to determine if DNA differences from person to person affect enhancer activity. The computer predicted that 43 percent of HARs contain two or more variants with large opposing effects: some variants in a given HAR made it a stronger enhancer, while other changes made the HAR a weaker enhancer. This result surprised the team, who had expected that all changes would push the enhancer in the same direction, or that some "hitchhiker" changes would have no impact on the enhancer at all. Measuring HAR strength To validate this compelling prediction, Pollard collaborated with the laboratories of Nadav Ahituv, Ph.D., and Alex Pollen, Ph.D., at UCSF. The researchers fused each HAR to a small DNA barcode. Each time a HAR was active, enhancing the expression of a gene, the barcode was transcribed into a piece of RNA. Then, the researchers used RNA sequencing technology to analyze how much of that barcode was present in any cell—indicating how active the HAR had been in that cell. "This method is much more quantitative because we have exact barcode counts instead of microscopy images," says Ahituv. "It's also much higher throughput; we can look at hundreds of HARs in a single experiment." When the group carried out their lab experiments on over 700 HARs in precursors to human and chimpanzee brain cells, the data mimicked what the machine learning algorithms had predicted. "We might not have discovered human HAR variants with opposing effects at all if the machine learning model hadn't produced these startling predictions," said Pollard. Implications for understanding psychiatric disease The idea that HAR variants played tug-of-war over enhancer levels fits in well with a theory that has already been proposed about human evolution: that the advanced cognition in our species is also what has given us psychiatric diseases. "What this kind of pattern indicates is something called compensatory evolution," says Pollard. "A large change was made in an enhancer, but maybe it was too much and led to harmful side effects, so the change was tuned back down over time—that's why we see opposing effects." If initial changes to HARs led to increased cognition, perhaps subsequent compensatory changes helped tune back down the risk of psychiatric diseases, Pollard speculates. Her data, she adds, can't directly prove or disprove that idea. But in the future, a better understanding of how HARs contribute to psychiatric disease could not only shed light on evolution, but on new treatments for these diseases. "We can never wind the clock back and know exactly what happened in evolution," says Pollard. "But we can use all these scientific techniques to simulate what might have happened and identify which DNA changes are most likely to explain unique aspects of the human brain, including its propensity for psychiatric disease." More information: Sean Whalen et al, Machine learning dissection of human accelerated regions in primate neurodevelopment, Neuron (2023). DOI: 10.1016/j.neuron.2022.12.026 |
|
|
|
Post by Admin on Jan 29, 2023 18:43:12 GMT
The movement of people across the Bering Sea from North Asia to North America is a well-known phenomenon in early human history. Nevertheless, the genetic makeup of the people who lived in North Asia during this time has remained mysterious due to a limited number of ancient genomes analyzed from this region. Now, researchers reporting in Current Biology on January 12 describe genomes from ten individuals up to 7,500 years old that help to fill the gap and show geneflow from people moving in the opposite direction from North America to North Asia. Their analysis reveals a previously undescribed group of early Holocene Siberian people that lived in the Neolithic Altai-Sayan region, near to where Russia, China, Mongolia, and Kazakhstan come together. The genetic data show they were descendants of both paleo-Siberian and Ancient North Eurasian (ANE) people. "We describe a previously unknown hunter-gatherer population in the Altai as early as 7,500 years old, which is a mixture between two distinct groups that lived in Siberia during the last Ice Age," says Cosimo Posth at the University of Tübingen, Germany, and senior author of the study. "The Altai hunter-gatherer group contributed to many contemporaneous and subsequent populations across North Asia, showing how great the mobility of those foraging communities was." Posth notes that the Altai region is known in the media as the location where a new archaic hominin group, the Denisovans, was discovered. But the region also has importance in human history as a crossroad for population movements between northern Siberia, Central Asia, and East Asia over millennia. Posth and colleagues report that the unique gene pool they uncovered may represent an optimal source for the inferred ANE-related population that contributed to Bronze Age groups from North and Inner Asia, such as Lake Baikal hunter-gatherers, Okunevo-associated pastoralists, and Tarim Basin mummies. They uncovered Ancient Northeast Asian (ANA) ancestry as well -- which had initially been described in Neolithic hunter-gatherers from the Russian Far East -- in another Neolithic Altai-Sayan individual associated with distinct cultural features. The findings reveal the spread of ANA ancestry about 1,500 kilometers farther to the west than previously observed. In the Russian Far East, they also identified 7,000-year-old individuals with Jomon-associated ancestry, indicating links with hunter-gatherer groups from the Japanese Archipelago. The data also are consistent with multiple phases of gene flow from North America to northeastern Asia over the last 5,000 years, reaching the Kamchatka Peninsula and central Siberia. The researchers note that the findings highlight a largely interconnected population throughout North Asia from the early Holocene onwards. "The finding that surprised me the most is from an individual dated to a similar period as the other Altai hunter-gatherers but with a completely different genetic profile, showing genetic affinities to populations located in the Russian Far East," says Ke Wang at Fudan University, China, and lead author of the study. "Interestingly, the Nizhnetytkesken individual was found in a cave containing rich burial goods with a religious costume and objects interpreted as possible representation of shamanism." Wang says the finding implies that individuals with very different profiles and backgrounds were living in the same region around the same time. "It is not clear if the Nizhnetytkesken individual came from far away or the population from which he derived was located close by," she says. "However, his grave goods appear different than other local archeological contexts implying mobility of both culturally and genetically diverse individuals into the Altai region." The genetic data from the Altai show that North Asia harbored highly connected groups as early as 10,000 years ago, across long geographic distances. "This suggests that human migrations and admixtures were the norm and not the exception also for ancient hunter-gatherer societies," Posth says. Journal Reference: Ke Wang, He Yu, Rita Radzevičiūtė, Yuriy F. Kiryushin, Alexey A. Tishkin, Yaroslav V. Frolov, Nadezhda F. Stepanova, Kirill Yu. Kiryushin, Artur L. Kungurov, Svetlana V. Shnaider, Svetlana S. Tur, Mikhail P. Tiunov, Alisa V. Zubova, Maria Pevzner, Timur Karimov, Alexandra Buzhilova, Viviane Slon, Choongwon Jeong, Johannes Krause, Cosimo Posth. Middle Holocene Siberian genomes reveal highly connected gene pools throughout North Asia. Current Biology, 2023; DOI: 10.1016/j.cub.2022.11.062 Highlights •A distinctive Middle Holocene Siberian ancestry is found in Altai hunter-gatherers •It results from a mixture of paleo-Siberian and ancient North Eurasian ancestries •A contemporaneous Altai individual carries ancient Northeast Asian ancestry •Northeastern Siberians experienced a prolonged Native American-related geneflow Summary The peopling history of North Asia remains largely unexplored due to the limited number of ancient genomes analyzed from this region. Here, we report genome-wide data of ten individuals dated to as early as 7,500 years before present from three regions in North Asia, namely Altai-Sayan, Russian Far East, and the Kamchatka Peninsula. Our analysis reveals a previously undescribed Middle Holocene Siberian gene pool in Neolithic Altai-Sayan hunter-gatherers as a genetic mixture between paleo-Siberian and ancient North Eurasian (ANE) ancestries. This distinctive gene pool represents an optimal source for the inferred ANE-related population that contributed to Bronze Age groups from North and Inner Asia, such as Lake Baikal hunter-gatherers, Okunevo-associated pastoralists, and possibly Tarim Basin populations. We find the presence of ancient Northeast Asian (ANA) ancestry—initially described in Neolithic groups from the Russian Far East—in another Neolithic Altai-Sayan individual associated with different cultural features, revealing the spread of ANA ancestry ∼1,500 km further to the west than previously observed. In the Russian Far East, we identify 7,000-year-old individuals that carry Jomon-associated ancestry indicating genetic links with hunter-gatherers in the Japanese archipelago. We also report multiple phases of Native American-related gene flow into northeastern Asia over the past 5,000 years, reaching the Kamchatka Peninsula and central Siberia. Our findings highlight largely interconnected population dynamics throughout North Asia from the Early Holocene onward. www.sciencedirect.com/science/article/abs/pii/S0960982222018929 |
|
|
|
Post by Admin on Feb 16, 2023 4:10:50 GMT
NEW YORK – Ancient DNA sequences from a Late Pleistocene female found in Southwest China suggest that the region was home to distinct, relatively diverse populations, including a lineage related to present-day populations in East Asia and elsewhere and to Indigenous populations that first settled the Americas.  In a paper appearing in Current Biology on Thursday, researchers from the Chinese Academy of Sciences, the Yunnan Institute of Cultural Relics and Archaeology, and other centers used in-solution DNA capture and shotgun sequencing to put together a mitochondrial genome and a low-coverage nuclear genome from remains found in a cave in China's Yunnan province. The sample came from an ancient female known as MZR who lived during the Late Pleistocene period roughly 14,000 to 14,700 years ago, the authors explained. A series of previous anthropological analyses suggested that MZR had physical and morphological features resembling both anatomically modern humans (AMH) and archaic hominins, including Neanderthal-like skull features. The new ancient genomic sequence data "confirm that [MZR] possesses diverse genetic components and represents an early diversified population, suggesting the scenarios of more diverse AMH lineages than previously thought during the Late Pleistocene in southern East Asia," senior and co-corresponding author Bing Su, a researcher with the Chinese Academy of Sciences' Kunming Institute of Zoology, and his colleagues explained, noting that the work "paves the way to explore genetic explanations of morphological complexities of early hominins."  By utilizing the published aDNA data, we reconstructed the spatial-temporal distribution of an East Asian-specific variant (OCA2-His615Arg) that contributes to skin lightening due to local Darwinian positive selection (Figure 5, left panel). [60][61][62][63] It turned out that all the Late Pleistocene individuals (e.g., MZR, Tianyuan, Amur-33K, Amur-19K, and UKY) lack the derived allele (OCA2-615Arg). The first presence of the adaptive allele (OCA2-615Arg) was in Liangdao 2-7.5 kya from coastal southern China, 39 and it quickly elevated to medium frequency (25.67%, 29/113), mainly in coastal East Asia, and then spread to northern East Asia 3,500 years ago, and finally became dominant (60.00%) in current East Asians (Figure 5, left panel; Data S1N). After extracting ancient DNA from a cranium bone sample, the investigators relied on modified human whole-genome probes to nab human-like sequences for subsequent amplification and library preparation steps. With dozens of DNA libraries, they ultimately generated hundreds of millions of sequence reads, providing a low-coverage look at the nuclear genome and a mitochondrial genome covered at an average depth of roughly 125-fold. Despite the archaic hominin-like features found in the ancient individual and the presence of archaic sequences introgressed from Denisovans and Neanderthals, the team's analyses indicated that MZR was part of an "early diversified" AMH lineage with genetic ties to existing populations. When they considered the ancient genome in the context of sequences from ancient and contemporary individuals from global populations, the investigators found that MZR carried ancestry from southern portions of East Asia that is related to that reported in Native Americans, consistent with the notion that migrations across the Bering Strait involved individuals migrating to Siberia from the southern reaches of East Asia.  "Our results indicate that MZR is a modern human who represents an early diversified lineage in East Asia," the authors reported, noting that "MZR is linked deeply to the East Asian ancestry that contributed to First Americans." On the mitochondrial DNA side, meanwhile, the researchers saw signs that MZR belonged to a maternal lineage that appeared to be basal to an M9 mitochondrial haplogroup with present-day offshoots believed to have moved south to north in mainland East Asia up to 28,000 years ago and into Southeast Asia, the Solomon Islands, and beyond. "[T]he discovery of an extinct basal M9 lineage for the MZR [individual] suggests a rich matrilineal diversity of human populations in southern East Asia during the Late Pleistocene," the authors explained. Together, the study's authors suggested, the new genomic insights support the notion that ancient populations from southern Asian sites were genetically nuanced, with more distinct genetic features than those found at more northerly sites in East or Southeast Asia.  The team reportedly expects to sequence still more ancient individuals from southern China and other parts of East Asia, including individuals from populations appearing prior to those identified in the Maludong, or Red Deer, Cave where MZR's remains were discovered. "Such data will not only help us paint a more complete picture of how our ancestors migrated but also contain important information about how humans change their physical appearance by adapting to local environments over time," Su said in a statement. A Late Pleistocene human genome from Southwest China Summary Southern East Asia is the dispersal center regarding the prehistoric settlement and migrations of modern humans in Asia-Pacific regions. However, the settlement pattern and population structure of paleolithic humans in this region remain elusive, and ancient DNA can provide direct information. Here, we sequenced the genome of a Late Pleistocene hominin (MZR), dated ∼14.0 thousand years ago from Red Deer Cave located in Southwest China, which was previously reported possessing mosaic features of modern and archaic hominins. MZR is the first Late Pleistocene genome from southern East Asia. Our results indicate that MZR is a modern human who represents an early diversified lineage in East Asia. The mtDNA of MZR belongs to an extinct basal lineage of the M9 haplogroup, reflecting a rich matrilineal diversity in southern East Asia during the Late Pleistocene. Combined with the published data, we detected clear genetic stratification in ancient southern populations of East/Southeast Asia and some degree of south-versus-north divergency during the Late Pleistocene, and MZR was identified as a southern East Asian who exhibits genetic continuity to present day populations. Markedly, MZR is linked deeply to the East Asian ancestry that contributed to First Americans. www.sciencedirect.com/science/article/abs/pii/S0960982222009289 |
|
|
|
Post by Admin on Feb 18, 2023 19:12:07 GMT
 A group of anthropologists had hired a team of local Thai rubber tree farmers to roam the hills in search of the nomadic hunter gatherers, but no one had been able to locate them. Back in the village, Austrian evolutionary anthropologist Tobias Göllner waited impatiently with his two colleagues, Helmut Lukas and Helmut Schaschl. If they could not make contact with the Maniq in the next week, they would have to return to Vienna empty-handed. Just as the academics were beginning to lose hope, one of the planters returned with good news: He had made contact with Ay-Kai, the “speaker” of a Maniq family group who Lukas had befriended over years of fieldwork. Ay-Kai’s family group was waiting for the researchers nearby. Göllner, the two Helmuts and a Thai linguist named Pacchira Chindaritha quickly packed up their things and hired a jeep to drop them off at the end of a rutted-out farm road. When they got there, the Maniq had erected a camp under the shade of a large limestone cliff. One sleeping platform with a thatched roof made from sticks and fresh leaves was set aside for the visitors. For the first few days, the adult Maniq mostly ignored Göllner’s presence. Ay-Kai, Lukas and Chindaritha caught up with each other during idle moments, but Göllner spent most of his time with the youngsters. “The children are not afraid of you. They’re very curious,” Göllner says. “From their perspective, I’m not really good at anything. They like teaching you stuff.” Finally, on the researcher’s last day at the camp, Ay-Kai asked Lukas the obvious question. Why had this young, seemingly-frail Austrian come all this way to visit them? With Chindaritha translating, Göllner did his best to explain his motivations. He wanted to trace the Maniq’s ancestry through genetic analysis. “You can’t really explain bifurcating trees to them from a genetics perspective,” Göllner says. “But you can use trees as a metaphor.” Göllner’s patience with the youngsters seemed to have paid off. The Maniq took pity on the strange young man and agreed to donate some saliva for later analysis. Three years later, in a paper in Genome Biology and Evolution published in April, Göllner and his colleagues revealed the most detailed genetic analysis to-date of the primarily hunter-gatherer society. Ancient Origins For millennia, the Maniq people have subsisted on foraged plant foods and wild game in the forested hills on the Thai-Malay border. And though the Maniq have had increasing contact with the outside world in recent decades, little is known about their ancestry. Prior to Göllner’s study, most theories on the origin of the Maniq centered around a great migration that populated the Malay peninsula between 40,000 and 50,000 years ago. The theories posit that the Maniq’s ancestors, the hunter-gatherer Hòabìnhian society, were among the first humans to inhabit southern Thailand. If the Maniq are indeed descendents of this ancient migration, their genetic lineage might be closely linked to other diverse ethnic groups in isolated parts of Southeast Asia — including the Semang in Malaysia, certain Filipino islanders and the Andamanese people, Indigenous inhabitants of the Andaman and Nicobar Islands in the Bay of Bengal. Göllner’s analysis supports the Hòabìnhian theory. The Maniq seem to be very closely related to the Semang, an Indigenous group from lands just over the Malay border. But, importantly, Göllner found strong evidence that the Maniq had not actually mixed with these populations for thousands of years. “We have this huge genetic drift, which points to a very long period of isolation,” he says. “Unfortunately, the algorithm we use to estimate the length of that period isn’t very good with heavily drifted data. The estimation was around 50,000 years, but we couldn’t publish it because of how imprecise it was.” Most other ethnic groups that can trace their lineage back to Hòabìnhian ancestors have mixed with other East Asian populations in the modern era. Though Göllner’s analysis revealed the presence of DNA from other East Asian sources in the Maniq’s genetics, this influence seems to be relatively small, comparatively. In other words, the Maniq may be the most direct descendants of the hunter-gatherers that populated the Malay peninsula tens of thousands of years ago. Ancient Knowledge What Göllner found in the DNA of 11 Maniq individuals is mirrored in the society’s culture and traditions. For example, their language has ancient roots that make it difficult for native Thai speakers to learn. And while many Indigenous peoples in Southeast Asia have embraced agriculture, the Maniq still live as their ancestors did. They hunt wild meat, gather nuts and tubers and use medicinal herbs to treat illness. Essentially, the remote, densely forested hills of Southern Thailand have buffered the culture from outside influence. During the rise and fall of colonialism, the society retained was mostly left alone by outsiders. But global capitalism may prove a more persistent force of change. In recent decades, the Maniq’s territory has shrunk at the edges; neat rows of rubber trees have replaced once-wild forests. It is not uncommon for Maniq to barter their labor for clothing or food in the villages that spring up near these plantations. Influence, too, has come from the curious academics and tourists who come bearing gifts and ideas. Ultimately, though, Göllner hopes that these exchanges will prove beneficial to both sides. “Industrial societies are very expansive. Contact with the Maniq is inevitable — it will happen,” he argues. "It's hard to gauge exactly how much we are influencing them, though we try to minimize it. What I can estimate is how much they influenced us, both scientifically and personally." Though it’s probably too late for Göllner to become proficient as hunter or gatherer, he’s still learned a great deal from his time with the Maniq, whose knowledge is not limited to practical skills. Maniq culture has survived, in part, due to their skill in sustaining a community. This can be observed in simple practices, like those surrounding possessions, which are often regarded as communal resources. A shirt, for example, may be passed from one person to another as needed for trips to nearby village. At meal times, everyone must have food in front of them before anyone can begin to eat. The same goes for cigarettes. “You roll cigarettes until everyone has one. The last one is for you,” Göllner says. The Maniq are likely one of the oldest communal societies on Earth, and they may have something to teach the rest of us. Göllner hopes that his work will motivate people in Thailand and beyond to value and respect the Maniq. If he’s right, they might have a chance at sustaining their traditions for a little bit longer. “Hunter-gatherer populations are the most important source of insight that we can gain from humans,” Göllner says. “It’s the way that Homo sapiens lived for probably 280,000 years. We have to be aware that these practices still exist. If you have an idea in your head about human nature, the first thing you should do is see if it holds true in a hunter-gatherer context.” academic.oup.com/gbe/article/14/4/evac021/6526392 |
|
|
|
Post by Admin on Feb 22, 2023 17:19:12 GMT
People have inhabited the Andes mountains of South America for more than 9,000 years, adapting to the scarce oxygen available at high altitudes, along with cold temperatures and intense ultraviolet radiation. A new genomic study suggests that Indigenous populations in present-day Ecuador also adapted to the tuberculosis bacterium, thousands of years before the arrival of Europeans. The journal iScience published the findings, led by scientists at Emory University.  "We found that selection for genes involved in TB-response pathways started to uptick a little over 3,000 years ago," says Sophie Joseph, first author of the paper and an Emory graduate student in anthropology. "That's an interesting time because it was when agriculture began proliferating in the region. The development of agriculture leads to more densely populated societies that are better at spreading a respiratory pathogen like TB." The investigators had originally set out to investigate how the Indigenous people of Ecuador adapted to living at high altitude. "We were surprised to find that the strongest genetic signals of positive selection were not associated with high altitude but for the immune response to tuberculosis," says John Lindo, Emory assistant professor of anthropology and senior author of the study. "Our results bring up more questions regarding the prevalence of tuberculosis in the Andes prior to European contact." The Lindo lab specializes in mapping little-explored human lineages of the Americas. Previously published research found evidence of the tuberculosis bacterium in the skeletal material of 1,400-year-old Andean mummies, contradicting some theories that TB did not exist in South America until the arrival of Europeans 500 years ago. The current paper provides the first evidence for a human immune-system response to TB in ancient Andeans and gives clues to when and how their genomes may have adapted to that exposure. "Human-pathogen co-evolution is an understudied area that has a huge bearing on modern-day public health," Joseph says. "Understanding how pathogens and humans have been linked and affecting each other over time may give insights into novel treatments for any number of infectious diseases." Co-authors of the paper include scientists at Central University of Ecuador, Technical University of Manabi in Ecuador, University of Pavia in Italy, University of Iowa and Florida Atlantic University. The researchers sequenced whole genomes using blood samples from 15 present-day Indigenous individuals living at altitudes above 2,500 meters in several different Ecuadorian provinces. They performed a series of scans to look for signatures of positive selection for genes in their ancestral past. "Computational techniques for sequencing genomes and modeling ancestral selection keep improving," Joseph says. "The genomes of people living today give us a window into the past." Among the strongest signals detected were for biomarkers that are switched on in modern humans during an active TB infection. The researchers modeled the timing of selection for several of the genes involved in the TB-response pathways. Although they were not as strong as for exposure to TB, some signals were also detected for biomarkers related to adaptation to hypoxia, or low levels of oxygen in the blood that result from living at high altitude. Previous research has revealed stark differences in how high-altitude populations in Tibet, Ethiopia and the Peruvian Andes adapted to hypoxia. "For the Ecuadorean samples, we did see a couple of overlaps with studies from the Peruvian Andes in the overarching genes involved in the selection for hypoxia, although the variants were slightly different," Joseph says. "To me, that suggests that there may have been independent adaptations within even small populations, at the community level. It shows the robustness of the genome to solve adaptive problems through different pathways." Joseph plans a career focused on mapping ancestral data for Indigenous populations from the Americas. "South America has far fewer genomic studies and publications compared to Europe and I'd like to help close that gap," she says. "I want to understand human evolution and health from an integrated biological perspective," Joseph adds. "The genome can reveal many fascinating things and yet it is just one aspect of a human being. You also have to consider the environment and social-cultural aspects." More information: Sophie K. Joseph et al, Genomic evidence for adaptation to tuberculosis in the Andes before European contact, iScience (2023). DOI: 10.1016/j.isci.2023.106034 www.cell.com/iscience/fulltext/S2589-0042(23)00111-6 |
|

