|
|
Post by Admin on May 9, 2023 18:23:04 GMT
Ancient human DNA recovered from a Palaeolithic pendant Abstract Artefacts made from stones, bones and teeth are fundamental to our understanding of human subsistence strategies, behaviour and culture in the Pleistocene. Although these resources are plentiful, it is impossible to associate artefacts to specific human individuals1 who can be morphologically or genetically characterized, unless they are found within burials, which are rare in this time period. Thus, our ability to discern the societal roles of Pleistocene individuals based on their biological sex or genetic ancestry is limited2,3,4,5. Here we report the development of a non-destructive method for the gradual release of DNA trapped in ancient bone and tooth artefacts. Application of the method to an Upper Palaeolithic deer tooth pendant from Denisova Cave, Russia, resulted in the recovery of ancient human and deer mitochondrial genomes, which allowed us to estimate the age of the pendant at approximately 19,000–25,000 years. Nuclear DNA analysis identifies the presumed maker or wearer of the pendant as a female individual with strong genetic affinities to a group of Ancient North Eurasian individuals who lived around the same time but were previously found only further east in Siberia. Our work redefines how cultural and genetic records can be linked in prehistoric archaeology. Main Palaeolithic assemblages typically contain a multitude of objects that may differ in age by hundreds or thousands of years, even when found in close proximity1. Thus, it can be challenging to associate human remains with specific objects. Recent advances in the retrieval of human DNA from sediments6,7,8 can be used to connect artefacts with genetic populations. However, precise identification of the specific makers or users of these objects would require the recovery of human DNA directly from the objects themselves, analogous to modern-day forensic investigations. In theory, such analyses are most promising for artefacts made from animal bones or teeth, not only because they are porous and thereby conducive to the penetration of body fluids (for example, sweat, blood or saliva) but also because they contain hydroxyapatite, which is known to adsorb DNA and reduce its degradation by hydrolysis and nuclease activity9,10. Ancient bones and teeth may therefore function as a trap not only for DNA that is released within an organism during its lifetime and subsequent decomposition but also for exogenous DNA that enters the matrix post-mortem through microbial colonization11 or handling by humans. However, DNA extraction from ancient skeletal material either requires destructive sampling, or risks alteration of specimens if they are directly submerged in extraction buffer12,13. Conservation is a primary concern because of the scarcity of bone and tooth artefacts at Pleistocene sites, especially of pendants and other ornaments that were extensively handled or worn in close body contact. We therefore set out to develop a method for DNA isolation from bones and teeth that preserves the integrity of the material, including surface microtopography, and to investigate the possibility of DNA retrieval from bone and tooth artefacts. A non-destructive DNA isolation method To identify reagents compatible with non-destructive DNA extraction, we selected ten unmodified faunal remains from the Palaeolithic sites of Quinçay and Les Cottés in France (Extended Data Table 1 and Supplementary Information 1), which were similar in size and shape to material typically used for osseous artefact production, and submerged them in several reagents previously used in ancient DNA extraction, as well as in water for comparison. These included (1) a guanidinium thiocyanate-containing reagent previously suggested for non-destructive DNA extraction12, (2) an ethylenediaminetetraacetate (EDTA) solution, which is a decalcifier commonly used in ancient DNA extraction14,15,16, (3) a sodium hypochlorite (bleach) solution, which is an oxidizing reagent used to remove surface-exposed contaminant DNA11,17, and (4) a sodium phosphate buffer supplemented with detergent11, which has been recently shown to enable temperature-controlled DNA release from powdered bone samples18. Mapping of the microtopography using quantitative 3D surface texture analysis19,20 before and after the treatments revealed substantial surface alterations on all objects exposed to either the guanidinium thiocyanate reagent or EDTA (Extended Data Fig. 1 and Supplementary Information 2). By contrast, only sporadic and smaller alterations were detected with the other reagents, including sodium phosphate buffer (Fig. 1b), possibly due to the removal of traces of sediment and other small particles, as indicated by visible changes in coloration of some of the objects (Extended Data Fig. 2). On the basis of these results, we developed a non-destructive DNA isolation method for the stepwise release of DNA from the bone or tooth matrix using serial incubations in sodium phosphate buffer at 21, 37, 60 and 90 °C, with three incubations per temperature (Fig. 1a). Fig. 1: Overview of the non-destructive DNA extraction method. 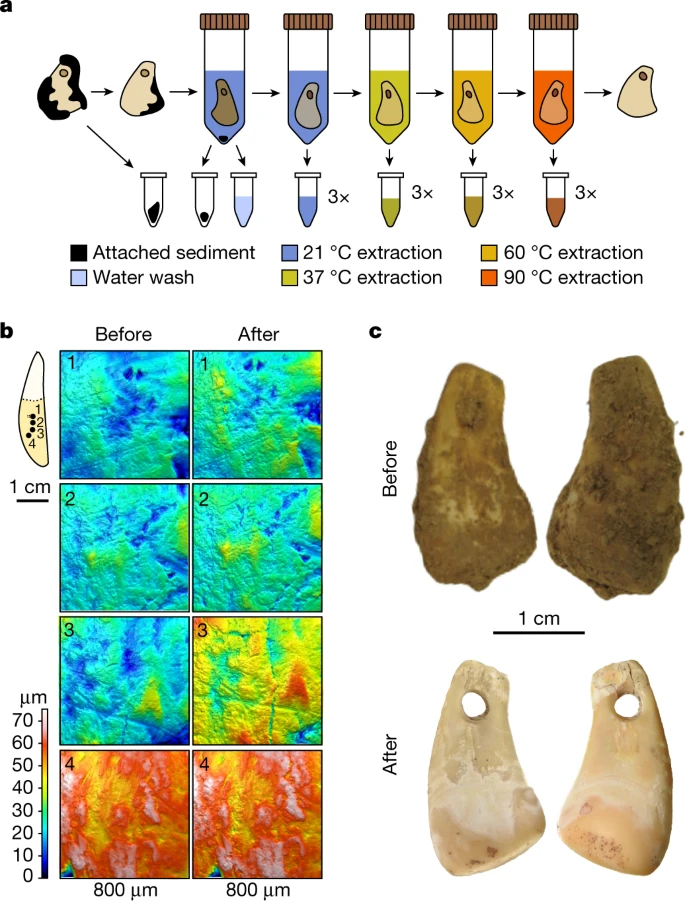 a, Workflow of the gradual, non-destructive DNA extraction method using sodium phosphate buffer at elevated temperatures. b, Four 3D surface texture measurements (1–4) indicated on the outline of a tooth used for testing (SP6649) before and after non-destructive DNA extraction showing no substantial surface alterations. c, Photographs of DCP1 before and after cleaning and non-destructive DNA extraction. www.nature.com/articles/s41586-023-06035-2 |
|
|
|
Post by Admin on Jun 5, 2023 19:32:45 GMT
A few years ago, Scottish woman Jo Cameron was found to be a medical marvel who felt next to no pain, fear or anxiety, and had faster wound healing, thanks to a specific gene mutation. Now, scientists have studied in more detail to figure out how this works, in the hopes of unlocking future drug targets. Cameron’s borderline superpower was only discovered in her mid-60s, after she underwent two major surgical procedures and reported little to no pain afterwards. When doctors followed up with her personal history with pain, she reported never really feeling minor cuts and scrapes, and some burns she didn’t even notice until she smelled burning flesh. She hadn’t needed painkillers after previous surgeries either. Cameron was referred to pain geneticists at Oxford and University College London, who identified two gene mutations as the root of her condition. One was in a gene called FAAH, which was previously known to control pain, mood and memory. The other was previously thought to be a non-functioning “junk” gene, but from this case was found to mediate the expression of FAAH. So they named it FAAH-OUT. In the new study, the team investigated how FAAH-OUT works biologically. Among their methods they used CRISPR gene editing on cells to check how the mutation affected other genes, and examined fibroblasts taken from other patients to study how FAAH and FAAH-OUT affect other molecular pathways.  It turns out that FAAH isn’t the only gene being turned down with these mutations – 348 others were also being suppressed, while an astonishing 797 genes were being turned up. Among them were the WNT pathway, which is linked to wound healing; BDNF, which is associated with mood regulation; and ACKR3, which regulates opioid levels. Altogether, these may help explain Cameron’s insensitivity to pain, her apparent wound healing speed boost, and her generally lower levels of anxiety and fear. It’s an intriguing case study, and along with others like the Marsili family, could help scientists identify new targets for medication to dampen pain or improve mental health symptoms. “The FAAH-OUT gene is just one small corner of a vast continent, which this study has begun to map,” said Dr. Andrei Okorokov, senior author of the study. “As well as the molecular basis for painlessness, these explorations have identified molecular pathways affecting wound healing and mood, all influenced by the FAAH-OUT mutation. As scientists it is our duty to explore and I think these findings will have important implications for areas of research such as wound healing, depression and more.” The research was published in the journal Brain. Source: UCL Abstract Chronic pain affects millions of people worldwide and new treatments are needed urgently. One way to identify novel analgesic strategies is to understand the biological dysfunctions that lead to human inherited pain insensitivity disorders. Here we report how the recently discovered brain and dorsal root ganglia-expressed FAAH-OUT long non-coding RNA (lncRNA) gene, which was found from studying a pain-insensitive patient with reduced anxiety and fast wound healing, regulates the adjacent key endocannabinoid system gene FAAH, which encodes the anandamide-degrading fatty acid amide hydrolase enzyme. We demonstrate that the disruption in FAAH-OUT lncRNA transcription leads to DNMT1-dependent DNA methylation within the FAAH promoter. In addition, FAAH-OUT contains a conserved regulatory element, FAAH-AMP, that acts as an enhancer for FAAH expression. academic.oup.com/brain/advance-article/doi/10.1093/brain/awad098/7169317 |
|
|
|
Post by Admin on Jan 4, 2024 20:17:48 GMT
The spatiotemporal patterns of major human admixture events during the European Holocene Recent studies have shown that population mixture (or ‘admixture’) is pervasive throughout human history, including mixture between the ancestors of modern humans and archaic hominins (i.e., Neanderthals and Denisovans), as well as in the history of many contemporary human groups such as African Americans, South Asians, and Europeans (Pickrell and Reich, 2014). Understanding the timing and signatures of admixture offers insights into the historical context in which the mixture occurred and enables the characterization of the evolutionary and functional impact of the gene flow. Many admixed groups are formed due to population movements involving ancient migrations that predate historical records. The recent availability of genomic data for a large number of present-day and ancient genomes provides an unprecedented opportunity to reconstruct population events using genetic data, providing evidence complementary to linguistics and archaeology. To characterize patterns of admixture, genetic methods use the insight that the genome of an admixed individual is a mosaic of chromosomal segments inherited from distinct ancestral populations (Chakraborty and Weiss, 1988). Due to recombination, these ancestral segments get shuffled in each generation and become smaller and smaller over time. The length of the segments is inversely proportional to the time elapsed since the mixture (Chakraborty and Weiss, 1988; Moorjani et al., 2011). Several genetic approaches – ROLLOFF (Moorjani et al., 2011; Patterson et al., 2012), ALDER (Loh et al., 2013), Globetrotter (Hellenthal et al., 2014), and Tracts (Gravel, 2012) – have been developed that use this insight by characterizing patterns of admixture linkage disequilibrium (LD) or haplotype lengths across the genome to infer the timing of mixture. Haplotype-based methods perform chromosome painting or local ancestry inference at each locus in the genome and characterize the distribution of ancestry tract lengths to estimate the time of mixture (Gravel, 2012; Hellenthal et al., 2014). This requires accurate phasing and inference of local ancestry, which is often difficult when the admixture events are old (as ancestry blocks become smaller over time) or when reference data from ancestral populations is unavailable. Admixture LD-based methods, on the other hand, measure the extent of the allelic correlation across markers to infer the time of admixture (Loh et al., 2013; Moorjani et al., 2011). They do not require phased data from the target or reference populations and work reliably for dating older admixture events (>100 generations). However, they tend to be less efficient in characterizing admixture events between closely related ancestral groups. While highly accurate for dating admixture events using data from present-day samples, current methods do not work reliably for dating admixture events using ancient genomes. Ancient DNA samples often have high rates of DNA degradation, contamination (from human and other sources), and low sequencing depth, leading to a large proportion of missing variants and uneven coverage across the genome (Orlando et al., 2021). Additionally, most studies generate pseudo-haploid genotype calls – consisting of a haploid genotype determined by randomly selecting one allele at the variant site – that can lead to some issues in the inference. In such sparse datasets, estimating admixture LD can be noisy and biased (see Simulations below). Moreover, haplotype-based methods require phased data from both admixed and reference populations which remains challenging for ancient DNA specimens (Gravel, 2012; Hellenthal et al., 2014). An extension of admixture LD-based methods, recently introduced by Moorjani et al., 2016, leverages ancestry covariance patterns that can be measured in a single sample using low coverage data. This approach measures the allelic correlation across neighboring sites, but instead of measuring admixture LD across multiple samples, it integrates data across markers within a single diploid genome. Using a set of ascertained markers that are informative for Neanderthal ancestry (where sub-Saharan Africans are fixed for the ancestral alleles and Neanderthals have a derived allele), Moorjani et al., 2016, inferred the timing of Neanderthal gene flow in Upper Paleolithic Eurasian samples and showed the approach works accurately in ancient DNA samples (Moorjani et al., 2016). However, this approach is inapplicable for dating admixture events within modern human populations, as there are very few fixed differences across populations (Auton et al., 2015). Motivated by the single sample statistic in Moorjani et al., 2016, we developed DATES (Distribution of Ancestry Tracts of Evolutionary Signals) that measures the ancestry covariance across the genome in a single admixed individual, weighted by the allele frequency difference between two ancestral populations. This method was first introduced in Narasimhan et al., 2019, where it was used to infer the date of gene flow between groups related to Ancient Ancestral South Indians, Iranian farmers, and Steppe pastoralists in ancient South and Central Asian populations (Narasimhan et al., 2019). In this study, we evaluate the performance of DATES by carrying out extensive simulations for a range of demographic scenarios and comparing the approach to other published genomic dating methods. We then apply DATES to infer the chronology of the genetic formation of the ancestral populations of Europeans and the spatiotemporal patterns of admixture during the European Holocene using data from ~1100 ancient DNA specimens spanning ~8000–350 BCE. elifesciences.org/articles/77625 |
|
|
|
Post by Admin on Jan 6, 2024 18:40:31 GMT
Overview of DATES: model and simulations DATES estimates the time of admixture by measuring the weighted ancestry covariance across the genome using data from a single diploid genome and two reference populations (representing the ancestral source populations). DATES works like haplotype-based methods as it is applicable to a single genome and not like admixture LD-based methods, which by definition require multiple genomes to be co-analyzed; but unlike haplotype-based methods, it is more flexible as it does not require local ancestry inference. There are three main steps in DATES: we start by first learning the genome-wide ancestry proportions by performing a simple regression analysis to model the observed genotypes in an admixed individual as a linear mix of allele frequencies from two reference populations. For each marker, we then compute the likelihood of the observed genotype in the admixed individual using the estimated ancestry proportions and allele frequencies in each reference population (this is similar in spirit to local ancestry inference). This information is, in turn, used to compute the joint likelihood of shared ancestry at two neighboring markers, accounting for the probability of recombination between the two markers. Finally, we compute the covariance across pairs of markers located at a particular genetic distance, weighted by the allele frequency differences in the reference populations (Materials and methods). Following Moorjani et al., 2016, we bin the markers that occur at a similar genetic distance across the genome, rather than estimating admixture LD for each pair of markers, and compute the covariance across increasing genetic distance between markers. The estimated covariance is expected to decay exponentially with genetic distance, and the rate of decay is informative of the time of the mixture (Chakraborty and Weiss, 1988; Moorjani et al., 2011). Assuming the gene flow occurred instantaneously, we can then infer the average date of gene flow by fitting an exponential distribution to the decay pattern (Materials and methods). In cases where data for multiple individuals is available, we compute the likelihood by summing over all individuals. To make DATES computationally tractable, we implement the fast Fourier transform (FFT) for calculating ancestry covariance as described in ALDER (Loh et al., 2013). This provides a speedup from O(n2) to O(nlogn) that reduces the typical runtimes from hours to seconds with minimal loss in accuracy (Appendix 1—figure 2). To assess the reliability of DATES, we performed simulations where we constructed 10 admixed diploid genomes by randomly sampling haplotypes from two source populations (Materials and methods). Briefly, we simulated individual genomes with 20% European and 80% African ancestry by using phased haplotypes of northern Europeans (Utah European Americans, CEU) and west Africans (Yoruba from Nigeria, YRI) from the 1000 Genomes Project, respectively (Auton et al., 2015). As reference populations in DATES, we used closely related surrogate populations of French and Yoruba respectively, from the Human Genome Diversity Panel (HGDP) (Li et al., 2008). We first investigated the accuracy of DATES by varying the time of admixture between 10 and 300 generations. For comparison, we also applied ALDER (Loh et al., 2013) to these simulations. Both methods reliably recovered the time of admixture up to 200 generations or ~5600 years ago, assuming a generation time of 28 years (Moorjani et al., 2016), though DATES was more precise than ALDER for older admixture events (>100 generations) (Figure 1—figure supplement 1, Appendix 1—table 4). Further, DATES shows accurate results even for single target samples (Figure 1A, Figure 1—figure supplement 2A) and even when few reference individuals are available for dating (Figure 1A, Figure 1—figure supplement 2B). However, the use of large numbers of reference samples, if available, can improve the inference. In DATES, allele frequencies of the reference populations are used for computing the likelihood as well as the weighted pairwise ancestry covariance across the genome (Materials and methods). With large samples, allele frequencies of the reference populations are more reliably computed, which in turn, can improve the precision of inferred dates (Figure 1A, Figure 1—figure supplement 2B). Figure 1 with 9 supplements 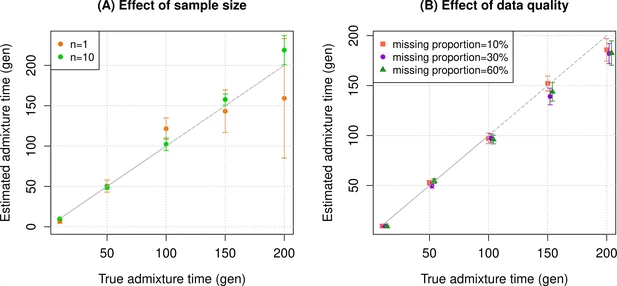 Simulation results. We constructed n admixed individuals with 20% European (CEU) and 80% African (YRI) ancestry using ~380,000 genome-wide SNPs for admixture dates ranging between 10 and 200 generations. To minimize any issues with overfitting, we used French and Yoruba from the Human Genome Diversity Panel as reference populations in DATES (Distribution of Ancestry Tracts of Evolutionary Signals). We show the true time of admixture (X-axis, in generations) and the estimated time of admixture (±1 SE) (Y-axis, in generations). Standard errors were calculated using a weighted block jackknife approach by removing one chromosome in each run (Materials and methods). (A) Effect of sample size: We varied the sample size (n) of target group between 1 and 10 individuals. (B) Effect of data quality: To mimic the features of ancient genomes, we generated n=10 target individuals with pseudo-haploid genotypes and missing genotype rate as 10% (orange), 30% (purple), and 60% (green). See Figure 1—figure supplements 1–9 for additional simulations to test the performance of DATES. R code to replicate this figure is available at: github.com/manjushachintalapati/DATES_EuropeanHolocene/blob/main/1.R. |
|
|
|
Post by Admin on Jan 8, 2024 21:18:53 GMT
Next, we tested DATES for features such as varying admixture proportions and use of surrogate populations as reference groups. By varying of European ancestry proportion between ~1% and 50% (the rest derived from west Africans), we observed DATES accurately estimated the timing in all cases (Figure 1—figure supplement 3A). However, the inferred admixture proportion was overestimated for lower admixture proportions (<10%) (Figure 1—figure supplement 3B). Thus, we caution against using DATES for estimating ancestry proportions. DATES works reliably for dating admixtures between related groups such as Europeans and Mexicans (FST~ 0.03), though it was unable to distinguish mixtures of Southern and Northern Europeans (FST< 0.005) (Figure 1—figure supplement 5).
We found DATES is robust to the use of highly divergent surrogates as reference populations. For example, the use of Khomani San as the reference population instead of the true ancestral population of Yoruba (FST ~ 0.1) provides unbiased dates of admixture (Figure 1—figure supplement 4). In this regard, for ancient DNA where sometimes only sparse data is available, one can also use present-day samples as reference populations to increase the quality and sample size of the ancestral groups. In principle, as long as the allele frequencies in the reference samples are correlated to the ancestral allele frequencies, the inference of admixture dates should remain unbiased (Materials and methods). In practice, however, recent demographic events (e.g., strong founder events or admixture from additional sources, etc.) in the history of the present-day samples could lead to significant deviation from the ancestral allele frequencies. Thus, the reference populations should be carefully chosen.
Another idea is to use the admixed populations themselves as one of the reference populations as demonstrated by the single reference setup in ALDER (Loh et al., 2013). Admixed individuals have intermediate allele frequencies to the ancestral populations and thus weighted LD or ancestry covariance can be computed with only one reference population (albeit, with reduced power). Loh et al., 2013, showed that the use of admixed populations as one of the references does not bias the rate of decay of the weighted LD (i.e., time of admixture), though the amplitude of the decay curve (not used in DATES) can be biased under some scenarios. To verify DATES provides reliable results under this setup, we applied DATES with a single reference population and used the admixed population as the other reference. Like ALDER, our inferred dates of admixture were accurate and comparable to using two reference populations. (Figure 1—figure supplement 6).
An important feature of DATES is that it does not require phased data and is applicable to datasets with small sample sizes, making it in principle useful for ancient DNA applications. To test the reliability of DATES for ancient genomes, we simulated data mimicking the relevant features of ancient genomes, namely small sample sizes (n=1–20), large proportions of missing genotypes (between 10% and 60%), and pseudo-haploid genotype calls (instead of diploid genotype calls) in reference and/or target samples. DATES showed reliable results under various setups, even when only a single admixed individual was available (Figure 1B, Figure 1—figure supplements 1–9). In contrast, admixture LD-based methods require more than one sample and do not work reliably with missing data. For example, ALDER estimates were very unstable for simulations with >40% missing data. For older dates (>100 generations), there was a slight bias even with >10% missing genotypes (Appendix 1—figure 5). This is expected as LD calculations leverage shared patterns across samples, thus variable missingness of genotypes across individuals leads to substantial loss of data leading to unstable and noisy inference. We also generated data for combinations of features including small sample sizes, pseudo-haploid genotypes with large proportions of missing genotypes in both target and reference samples, and use of highly divergent reference samples. We found DATES yielded reliable results with large amounts (~40–60%) of missing data, either in the target or references, even with highly divergent reference populations (Figure 1—figure supplement 8). This was also true when a single target sample was available, though as expected, the inference becomes noisier for older dates and large fractions of missing data (Figure 1—figure supplement 9). The robust performance of DATES in sparse datasets highlights a major advantage for ancient DNA applications.
DATES assumes a model of instantaneous gene flow with a single pulse of mixture between two source populations. However, many human populations have a history of multiple pulses of gene flow. To test the performance of DATES for multi-way admixture events, we generated admixed individuals with ancestry from three sources (East Asians, Africans, and Europeans) where the gene flow occurred at two distinct time points (Appendix 2—figure 1). By applying DATES with pairs of reference populations, we observed that DATES recovered both admixture times for target populations that had equal contributions from all three ancestral groups (Appendix 2—figure 2). In the case of unequal admixture proportions from three ancestral groups, DATES inferred the timing of the recent admixture event in most cases. In some cases, however, the inferred dates were intermediate to the two pulses when the ancestry proportion of the recent event was low (Appendix 2—figure 3). This confounding could be eliminated if the reference populations were set up to match the model of gene flow. For example, the inferred times of admixture were accurate if the two references used in DATES were: reference 1: the source population for the recent event and reference 2: pooled individuals from both ancestral populations that contributed to the first admixture event, or the intermediate admixed group formed after the first event (Appendix 2—table 1). This highlights how the choice of reference populations can help to tune the method to infer the timing of specific admixture events more reliably.
Finally, we explored the impact of more complex demographic events, including continuous admixture and founder events using coalescent simulations (Appendix 2). In the case of continuous admixture, DATES inferred an intermediate timing between the start and the end of the gene flow period, similar to other methods like ALDER and Globetrotter (Hellenthal et al., 2014; Loh et al., 2013; Appendix 2—table 2). In the case of populations with founder events, we inferred unbiased dates of admixture in most cases except when the founder event was extreme (Ne ~ 10) or the population had maintained a low population size (Ne < 100) until the present (i.e., no recovery bottleneck) (Appendix 2—figure 4, Appendix 2—table 3). In humans, few populations have such extreme founder events, and thus, in most other cases, our inferred admixture dates should be robust to founder events (Tournebize et al., 2020). We note that while DATES is not a formal test of admixture, in simulations, we find that in the absence of gene flow, the method does not infer significant dates of admixture even if the target has a complex demographic history (Appendix 2—figure 6, Appendix 2—figure 7).
|
|

