|
|
Post by Admin on May 19, 2014 22:19:40 GMT
Although Neanderthals are extinct, fragments of their genomes persist in contemporary humans. Here we show that while the genome-wide frequency of Neanderthal-like sites is approximately constant across all contemporary out-of-Africa populations, genes involved in lipid catabolism contain more than threefold excess of such sites in contemporary humans of European descent. Evolutionally, these genes show significant association with signatures of recent positive selection in the contemporary European, but not Asian or African populations. Functionally, the excess of Neanderthal-like sites in lipid catabolism genes can be linked with a greater divergence of lipid concentrations and enzyme expression levels within this pathway, seen in contemporary Europeans, but not in the other populations. We conclude that sequence variants that evolved in Neanderthals may have given a selective advantage to anatomically modern humans that settled in the same geographical areas. Neanderthal ancestry in contemporary human populationsWe searched for regional similarities to the Neanderthal genome in the genomes of 11 contemporary human populations, which have the best genome coverage in the 1,000 genomes project: three populations of African ancestry—HapMap African ancestry individuals from South West of the United States (ASW), Luhya individuals (LWK) and Yoruba individuals (YRI); three populations of East Asian ancestry—Han Chinese in Beijing (CHB), Han Chinese from South China (CHS) and Japanese (JPT); and five populations of European ancestry—CEPH individuals (CEU), HapMap Finnish individuals from Finland (FIN), British individuals from England and Scotland (GBR), Iberian populations in Spain (IBS) and Toscan individuals (TSI)13. For each pair of the human populations, the human genome sequences were compared with the Neanderthal and the chimpanzee genome sequences. We used the high-coverage Neanderthal genome sequence, obtained from the bone of one individual excavated in 2008 in the east gallery of Denisova Cave in the Altai mountains (Altai)14, 15, combined with the low-coverage Neanderthal genome sequence, obtained from three individuals (Vindjia)10, to reconstruct a consensus Neanderthal genome sequence by excluding all sites with sequence variation among individuals. We further used the reference chimpanzee genome in combination with genome sequence data from 10 chimpanzees16 to exclude sites variable among chimpanzees. Only single nucleotide sites displaying sequence differences between the chimpanzee16, 17 and the two reference Neanderthal genomes10, 15 were used in the analysis (n=1,158,559). Similar to Green et al.10, we used the D statistic to detect potential Neanderthal gene flow. For each pair of human populations, we defined two configurations for genomic sites: sites where the sequence of an individual from one human population (population A) matched the Neanderthal rather than chimpanzee genotype were assigned to the ABBA configuration; and sites where the sequence of the other human population (population B) matched the Neanderthal rather than chimpanzee genotype were assigned to the BABA configuration (Fig. 1a). For each population, the D statistic, which we will refer to as the fraction of Neanderthal-like sites (NLS), was calculated as the ratio of (#ABBA−#BABA) to (#ABBA+#BABA), where # stands for the number of genomic sites in the specific genotype configuration (ABBA or BABA; see Methods). While D-statistic values reflect relative similarity between the Neanderthal and the modern human genomes tested, they do not provide a quantitative estimate of Neanderthal ancestry, that is, a 5% D-statistic value reflects a higher similarity between population A and Neanderthal compared with that for population B, but does not signify a 5% level of Neanderthal ancestry in the population A genome.  Figure 1: Proportions of NLS in contemporary human populations.(a) Schematic representation of genomic distance calculations between contemporary human populations and Neanderthals. The genomes of out-of-Africa individuals were compared with the genomes of individuals of purely African ancestry (YRI). Single nucleotide differences from the Neanderthal genotype in an African genome were referred to as ‘ABBA’, while sites with the Neanderthal genotype in an out-of-Africa genome were referred to as ‘BABA’. (b) Average proportions of NLS in contemporary African (AF), European (EU) and Asian (AS) populations calculated based on sequence data from the 1,000 genomes project13; blue: genome wide (n=1,158,559 sites), red: LCP genes (n=498 sites). The error bars show the s.d. of the NLS proportion estimates. (c) Genomic distances between 11 contemporary human populations and Neanderthals; blue, genome wide; red, LCP genes. The maximal bar length corresponds to a NLS frequency of 30%. Placement of ASW and CEU individuals in sub-Saharan Africa and Western Europe, respectively, reflects their approximate historical geographical origins rather than their present location. Figure 1: Proportions of NLS in contemporary human populations.(a) Schematic representation of genomic distance calculations between contemporary human populations and Neanderthals. The genomes of out-of-Africa individuals were compared with the genomes of individuals of purely African ancestry (YRI). Single nucleotide differences from the Neanderthal genotype in an African genome were referred to as ‘ABBA’, while sites with the Neanderthal genotype in an out-of-Africa genome were referred to as ‘BABA’. (b) Average proportions of NLS in contemporary African (AF), European (EU) and Asian (AS) populations calculated based on sequence data from the 1,000 genomes project13; blue: genome wide (n=1,158,559 sites), red: LCP genes (n=498 sites). The error bars show the s.d. of the NLS proportion estimates. (c) Genomic distances between 11 contemporary human populations and Neanderthals; blue, genome wide; red, LCP genes. The maximal bar length corresponds to a NLS frequency of 30%. Placement of ASW and CEU individuals in sub-Saharan Africa and Western Europe, respectively, reflects their approximate historical geographical origins rather than their present location.In agreement with previous observations10, the genomes of contemporary humans of European and Asian descent showed greater similarities to the Neanderthal genome than did the genomes of the three populations of purely African descent. On average, the NLS frequency was 6.1±0.2% for contemporary humans of European and Asian descent, thus indicating a substantial excess of NLS in contemporary out-of-Africa populations (Fig. 1b,c, blue bars; Supplementary Tables 1–3). This D-statistic estimate is similar to the ones reported by other studies (4.8±0.2%)10, with the higher values obtained in our study potentially arising from the additional filtering of genomic sites polymorphic in Neanderthals. Further, in agreement with other studies10, there was no substantial difference in the genome-wide frequencies of NLS between European and Asian populations, with a slight tendency for higher frequencies in Asians: 5.9±0.08 and 6.2±0.06%, respectively18, 19. For each pair of human populations, we searched for the presence of functional groups of genes showing an unusual excess, or paucity, of NLS. This analysis, based on gene groups compiled according to gene ontology terms20, and conducted using the gene set enrichment analysis (GSEA) algorithm21, yielded an unexpected observation; we indeed find significant clustering of NLS in specific functional groups, but these functional groups differed substantially between contemporary European and Asian populations (Supplementary Data 1). Functional groups showing NLS enrichment in Asian populations mainly represent immune and haematopoietic pathways. The strongest signal of NLS enrichment was, however, observed in contemporary Europeans and included two functional groups: the lipid catabolic process (LCP) and its nested term—cellular LCP (GSEA, permutations P<0.01, significance score >3; Fig. 2a). Specifically, genes in the LCP term had the greatest excess of NLS in populations of European descent, with an average NLS frequency of 20.8±2.6% versus 5.9±0.08% genome wide (two-sided t-test, P<0.0001, n=379 Europeans and n=246 Africans; Fig. 1b,c, red bars; Supplementary Table 4; Supplementary Fig. 1). Further, among examined out-of-Africa human populations, the excess of NLS in LCP genes was only observed in individuals of European descent: the average NLS frequency in Asians is 6.7±0.7% in LCP genes versus 6.2±0.06% genome wide (Supplementary Table 4).  Figure 2: Outstanding genetic features of lipid catabolism genes.(a) Clustering of NLS in functional categories in the genomes of contemporary humans of European (EU) and Asian (AS) descent. The significance scores are proportional to the NES and inversely proportional to the false discovery rates calculated for each gene ontology (GO) term in the biological process category using the GSEA algorithm. Sizes of circles and squares represent NLS fractions in Europeans and Asians, respectively, for each GO term. Numbers near the circles mark the top two GO terms with a functional enrichment significance score greater than three, based on the distribution of Neanderthal-like genomic sites in Europeans: (1) LCP, (2) cellular LCP. (b) Positive selection signals in LCP gene regions estimated using CMS scores. Black squares represent the frequency of sites with elevated CMS scores (>1), potentially indicating genomic regions under recent positive selection in LCP gene regions, normalized by the frequency of such sites in all annotated genes within the same population. The boxplots show the variation of normalized site frequency estimates obtained by 1,000 bootstraps over LCP gene regions (n=38). The boxes show quartiles and the median of the data, the whiskers extend to the minimum and maximum data values located within 0.5 interquartile range from the box. The numbers above the boxplots show the proportion of bootstrap values where the normalized site frequency of elevated CMS scores in Africans (AF) or Asians (AS) was greater than, or equal to, the normalized site frequency in Europeans (EU). Figure 2: Outstanding genetic features of lipid catabolism genes.(a) Clustering of NLS in functional categories in the genomes of contemporary humans of European (EU) and Asian (AS) descent. The significance scores are proportional to the NES and inversely proportional to the false discovery rates calculated for each gene ontology (GO) term in the biological process category using the GSEA algorithm. Sizes of circles and squares represent NLS fractions in Europeans and Asians, respectively, for each GO term. Numbers near the circles mark the top two GO terms with a functional enrichment significance score greater than three, based on the distribution of Neanderthal-like genomic sites in Europeans: (1) LCP, (2) cellular LCP. (b) Positive selection signals in LCP gene regions estimated using CMS scores. Black squares represent the frequency of sites with elevated CMS scores (>1), potentially indicating genomic regions under recent positive selection in LCP gene regions, normalized by the frequency of such sites in all annotated genes within the same population. The boxplots show the variation of normalized site frequency estimates obtained by 1,000 bootstraps over LCP gene regions (n=38). The boxes show quartiles and the median of the data, the whiskers extend to the minimum and maximum data values located within 0.5 interquartile range from the box. The numbers above the boxplots show the proportion of bootstrap values where the normalized site frequency of elevated CMS scores in Africans (AF) or Asians (AS) was greater than, or equal to, the normalized site frequency in Europeans (EU).The excess of NLS observed in LCP genes for populations of European descent was based on a large number of sites (n=498), and was robust to bootstrapping across sites (P<0.01, 1,000 bootstraps, Supplementary Table 5; Supplementary Fig. 2). Notably, NLS were located in 23 independent genomic regions. Among the remaining 15 LCP genes that did not contain NLS, 8 did not contain sites showing divergence between Neanderthals and chimpanzees and the remaining 7 contained only a few such divergent sites (Supplementary Table 6). It is furthermore robust to the potential effects of DNA damage characteristic of ancient DNA samples, as excluding the C/T and A/G substitutions that may stem from deamination of cytosine residues in ancient DNA22, 23 did not affect the results (Supplementary Table 4). Repeating the analysis using the genome sequences of African (ASW, LWK, YRI), European (CEU, FIN, GBR, IBS) and East Asian (CHB, JPT) individuals, which were sequenced to deeper coverage at the pilot stage of the 1,000 genomes project, as well as the high-coverage genome sequences of African (ASW, LWK, YRI, MKK—Maasai in Kinyawa, Kenya), European (CEU, TSI) and East Asian (CHB, JPT) individuals provided by the Complete Genomics human diversity set24, confirmed our observations (Supplementary Tables 7 and 8). Finally, repeating the analysis using the high-coverage (Altai) and the low-coverage (Vindjia) Neanderthal genomes, separately, resulted in similar findings (Supplementary Table 9). The excess of NLS in LCP genes in the genomes of contemporary Europeans may be due to a rapid spread of Neanderthal alleles in European ancestors because of their adaptive significance. Specifically, one may hypothesize that, over time, Neanderthals acquired changes to lipid catabolism, which were beneficial for survival in the environmental conditions of prehistoric Europe and Central Asia. These adaptive variants may then have been acquired by the modern humans through introgression and rapidly brought to high frequency by positive selection. To test this hypothesis, we searched for signatures of positive selection in the genomes of contemporary humans of European, Asian and African decent using composite of multiple signals (CMS) scores25. High CMS values indicate genomic regions under recent positive selection based on three distinct signatures of selection: long-range haplotypes, differentiated alleles and high-frequency-derived alleles. We indeed found a significant excess of high CMS scores in the LCP gene regions of contemporary Europeans but not Asians or Africans (Fig. 2b). This effect was robust at different CMS score cutoffs and was specific to LCP: no significant excess of high CMS scores in individuals of European descent was observed in comparable genomic regions containing other metabolic genes (Supplementary Fig. 3). Furthermore, within the LCP term, high CMS scores found in contemporary Europeans were associated with genes containing the excess of NLS, but were not associated with other LCP genes (two-sample Wilcoxon test, P=0.0003; n=45 and 20; Supplementary Fig. 4). While we find changes in lipid catabolism particular to Europeans at the metabolite concentration and enzyme expression levels, the significance of these changes at the organismal level remains to be investigated. Still, the changes observed at the molecular level provide some clues. Among the seven metabolic categories associated with LCP, 2-lysophosphatidylcholine has been implicated in a number of functions, including reactive oxygen species generation, apoptotic and non-apoptotic death, as well as glucose-dependent insulin secretion32 (Fig. 4b,c). Furthermore, genetic variants linked to obesity (DAVID33, Fisher’s exact test, P<0.01 after multiple testing correction, n=38) hypertriglyceridemia and coronary heart disease, as well as triglycerides and cholesterol levels (DAVID, Fisher’s exact test, P<0.01 after multiple testing correction, n=38) in genome-wide association studies34 show a significant enrichment of LPC genes containing an excess of NLS (Supplementary Table 13). Notably, frequencies of these diseases have been shown to differ between individuals of European descent and other human populations35. These observations support a contribution of Neanderthal genetic variants to the phenotype of contemporary Europeans. Khrameeva, Ekaterina E., et al. " Neanderthal ancestry drives evolution of lipid catabolism in contemporary Europeans." Nature communications 5 (2014). |
|
|
|
Post by Admin on Aug 21, 2014 20:32:46 GMT
 Neanderthals and humans lived together in Europe for thousands of years, concludes a timeline based on radiocarbon dates from 40 key sites across Europe. The results, published today in Nature, may help to end a century-old deadlock over the demise of the Neanderthals and their relationship to humans. The researchers used 196 radiocarbon dates of organic remains to show that Neanderthals disappeared from Europe around 40,000 years ago, but still long after humans arrived in the continent. “Humans and Neanderthals were living contemporaneously for quite some period of time in different parts of Europe,” says Tom Higham, an archaeologist at the University of Oxford, UK, who led the study. The long overlap provided plenty of time for cultural exchange and interbreeding, he adds.  Exactly what happened 30,000–50,000 years ago still vexes archaeologists because the period is right at the limit of accurate radiocarbon dating. The technique is based on measuring the steady loss of radioactive carbon-14 molecules in organic remains. But after 30,000 years, 98% of the isotope is gone and younger carbon molecules are starting to infiltrate bones, making remains seem younger than they are. This means that dates for the final Neanderthals and for the first human occupations of Europe have been unreliable, fomenting the debate. But over the past decade, Higham and his team have developed techniques that provide more accurate readings in bones up to 55,000 years old (see Nature 485, 27–29; 2012). First, they use a chemical pretreatment to remove the contaminating carbon from the collagen in bones, then they measure the minuscule amounts of radiocarbon using a particle accelerator.  This animal bone with cut marks made by Neanderthals was found at Le Moustier in France The technique has allowed the researchers to redraft the prehistory of Europe cave by cave, and show that early humans arrived in southwestern England and Italy’s ‘heel’, for example, well over 40,000 years ago. They have now applied the technique to Neanderthal occupations across Europe, which are associated with stone tools known as Mousterian artifacts. From the Black Sea to the Atlantic coast of France, these artifacts and Neanderthal remains disappear from European sites at roughly the same time, 39,000–41,000 years ago, Higham’s team conclude. The data challenge arguments that Neanderthals endured in refuges in the southern Iberian Peninsula until as recently as 28,000 years ago. Humans, argues Higham’s team, were in Italy as early as 45,000 years ago, where they developed a stone-tool culture known as the Uluzzian industry. The team estimates that humans and Neanderthals overlapped for up to 5,400 years in parts of southern Europe, yet to a much lesser extent or not at all in other parts of the continent. “They were certainly in the same areas,” Higham says.  The coexistence also supports a controversial idea that some Neanderthal artifacts, such as shell beads and stone tools of the Châtelperronian industry that appeared in France and Spain more than 40,000 years ago, emerged from contact with humans, Higham says. “I absolutely agree with Tom,” says Paul Mellars, an archaeologist at the University of Cambridge, UK, who has long championed the idea that Neanderthals borrowed technologies from humans. “There would be opportunities for contacts or interactions thousands of times in most, if not all, areas of Europe,” he says.  Transitional site locations and Bayesian age ranges for the start and end of the Châtelperronian and Uluzzian technocomplexes. Meanwhile, Higham hopes that the timeline will address other mysteries surrounding Neanderthals, such as why they died out and how they interacted with humans. DNA recovered from remains in Europe and western Asia, for example, show that humans and Neanderthals interbred more than 50,000 years ago, probably as the common ancestor of Europeans and Asians emerged from Africa. There is still no evidence that humans and Neanderthals interbred while in Europe, but thousands of years of overlap makes sex more likely, Higham says. “I do like the idea that they aren’t really extinct and they do live on in us.”  Time slices for western Europe between 45,000 and 40,000 cal BP showing the distribution of the Mousterian, Châtelperronian and Uluzzian modelled ages. The timing of Neanderthal disappearance and the extent to which they overlapped with the earliest incoming anatomically modern humans (AMHs) in Eurasia are key questions in palaeoanthropology1, 2. Determining the spatiotemporal relationship between the two populations is crucial if we are to understand the processes, timing and reasons leading to the disappearance of Neanderthals and the likelihood of cultural and genetic exchange. Serious technical challenges, however, have hindered reliable dating of the period, as the radiocarbon method reaches its limit at ~50,000 years ago3. Here we apply improved accelerator mass spectrometry 14C techniques to construct robust chronologies from 40 key Mousterian and Neanderthal archaeological sites, ranging from Russia to Spain. Bayesian age modelling was used to generate probability distribution functions to determine the latest appearance date. We show that the Mousterian ended by 41,030–39,260 calibrated years BP (at 95.4% probability) across Europe. We also demonstrate that succeeding ‘transitional’ archaeological industries, one of which has been linked with Neanderthals (Châtelperronian)4, end at a similar time. Our data indicate that the disappearance of Neanderthals occurred at different times in different regions. Comparing the data with results obtained from the earliest dated AMH sites in Europe, associated with the Uluzzian technocomplex5, allows us to quantify the temporal overlap between the two human groups. The results reveal a significant overlap of 2,600–5,400 years (at 95.4% probability). This has important implications for models seeking to explain the cultural, technological and biological elements involved in the replacement of Neanderthals by AMHs. A mosaic of populations in Europe during the Middle to Upper Palaeolithic transition suggests that there was ample time for the transmission of cultural and symbolic behaviours, as well as possible genetic exchanges, between the two groups. Higham et al., The timing and spatiotemporal patterning of Neanderthal disappearance, Nature 485, 27–29; 2012. |
|
|
|
Post by Admin on Aug 22, 2014 19:57:55 GMT
Abstract Background: Genes and culture are believed to interact, but it has been difficult to find direct evidence for the process. One candidate example that has been put forward is lactase persistence in adulthood, i.e. the ability to continue digesting the milk sugar lactose after childhood, facilitating the consumption of raw milk. This genetic trait is believed to have evolved within a short time period and to be related with the emergence of sedentary agriculture. Results: Here we investigate the frequency of an allele (-13910*T) associated with lactase persistence in a Neolithic Scandinavian population. From the 14 individuals originally examined, 10 yielded reliable results. We find that the T allele frequency was very low (5%) in this Middle Neolithic hunter-gatherer population, and that the frequency is dramatically different from the extant Swedish population (74%). Conclusions: We conclude that this difference in frequency could not have arisen by genetic drift and is either due to selection or, more likely, replacement of hunter-gatherer populations by sedentary agriculturalists.  The ability to drink milk as an adult occurs at a high frequency in present-day populations that practice dairying and cattle rearing [1-4]. It has been suggested that this correlation represents a case of gene-culture co-evolution, i.e. an adaptive genetic trait exposed to positive selection induced by cultural practices. The -13910*T allele associated with this trait in Europeans appears to have been the target of strong selection over a relatively short period of time [5,6]. Such selection pressure could be one of several explanations for the high frequency of the derived allele (associated with allowing milk consumption in adulthood) in northern Europeans, the region where the allele is most common (74% in Sweden [7]). A single nucleotide polymorphism (SNP-13910 T/C), strongly associated with the ability to digest lactose in adulthood [8], has been used as a marker for the genetic trait. Notably, the allele frequency at the SNP and the extent of haplotype homozygosity around the particular SNP-allele indicate a history of a strong positive selection [5,6,9,10], and it has been suggested that this selective pressure was attributable to the introduction of agriculture and animal domestication [10,11]. The Pitted Ware Culture (PWC) was a major Neolithic hunter-gatherer population in Northern Europe and was partly contemporaneous with the farming TRB population (TRB after the German word Trichterbecherkultur, i.e Funnel Beaker Culture). The PWC are thought to have been present in Scandinavia between 5,400-4,300 years before present (BP) [12], which is later than the suggested initiation of selection for the T allele [6]. In this study, we find that the frequency of the derived allele is low in the PWC (5%) compared to the frequency in the extant Swedish population, and that the change in frequency is incompatible with genetic drift as the sole explanation under a model of population continuity. Thus, a genetic component interacting with culture, such as the ability to digest milk as an adult, could have been the result of the replacement of the hunter-gatherer population by an agricultural population. Alternatively, positive selection could have dramatically increased the frequency of the derived allele in the PWC, allowing for population continuity from the PWC to the extant Swedish population. 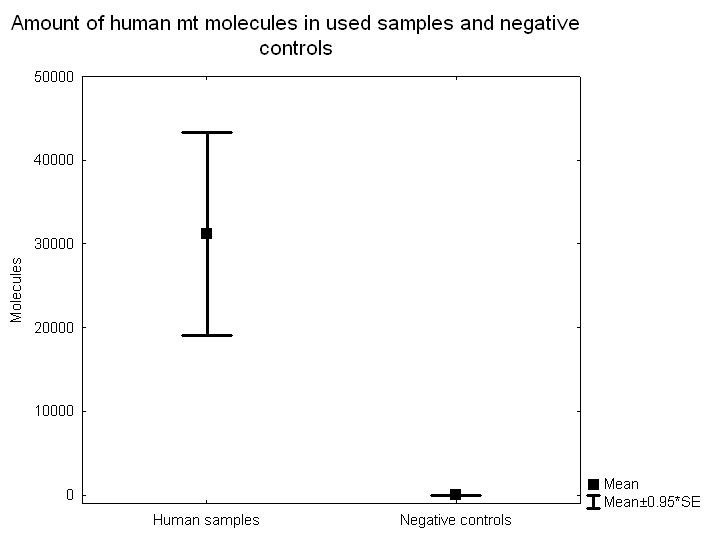 Figure 2. Real-time PCR quantitative data for the 10 samples and 98 negative controls, where 31 were nonhuman mammals and 67 were water controls or PCR controls. Figure 2. Real-time PCR quantitative data for the 10 samples and 98 negative controls, where 31 were nonhuman mammals and 67 were water controls or PCR controls.The material consists of duplicate samples (teeth and/or ulna, femur or fibula) from 14 prehistoric individuals (Table 1). The samples originate from four archaeological sites on Gotland in the Baltic Sea dating to the Middle Neolithic, 4,800-4,200 BP (Figure 1). The 14 samples, originally from a larger set of 36 from Gotland and mainland Sweden, were chosen on the basis that they had all yielded high amounts of mitochondrial DNA compared to negative controls (Figure 2) [13]. The samples were collected from the following sites on Gotland: Ajvide (n = 9) in the parish of Eksta, Visby town (n = 1), Ire (n = 2) in the parish of Hangvar and Fridtorp (n = 2) in the parish of Västerhejde. All samples are at least a 1,000 years younger than the earliest evidence of agriculture in Scandinavia. Hence they represent an exclusively non-agricultural lifestyle considerably older than agriculture in the area. All of the samples are from burial contexts and were excavated, handled and stored following standard protocols for the excavation and storage of archaeological material. Thus, no special precautions were taken to minimize human DNA contamination. The nonhuman faunal samples, used as negative controls, were subject to the same excavation and storage protocols as the human samples, and thus would not be expected to yield a different contamination pattern compared to the human samples. 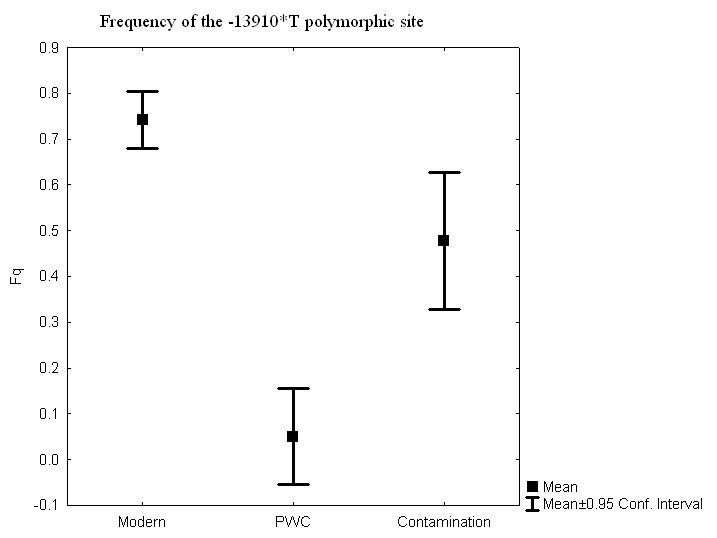 Figure 3. Frequency of the -13910*T polymorphic site in the lactase gene in three datasets: an extant Swedish population, a Swedish Neolithic hunter-gatherer population (PWC) and the negative controls. Figure 3. Frequency of the -13910*T polymorphic site in the lactase gene in three datasets: an extant Swedish population, a Swedish Neolithic hunter-gatherer population (PWC) and the negative controls.
The fourteen specimens all yielded replicable results and ten of these were successfully SNP-typed a minimum of four times (using both the 53 bp and the 168 bp fragment). Thus, even with thorough mitochondrial pre-selection, the success rate for the nuclear DNA sequences was down to 71%. As only one individual was heterozygote, it is not possible to calculate an allelic dropout rate, but from the four pyrosequencing results from the heterozygote individual, two replicate typing events expressed allelic dropout. A low number of positive results were detected in the negative controls, eight of the 59 Neolithic seals, seven of the 53 negative extraction controls and eight of the 47 PCR negative controls. In total, 14% of the negative controls were contaminated, and out of these, the frequency of the T allele was 52%. None of the contaminants could however be reproduced. The allele frequency in the negative controls was significantly different from the allele frequency in the ten archaeological samples used for further analysis (Fisher's Exact test, p < 0.001). The contamination frequency is in direct contrast to that of the short mitochondrial fragment, where 86% of the negative controls (98 negative controls, where 31 were nonhuman mammals and 67 water controls or PCR controls) expressed contamination, although to a more than 1000-fold lower quantity than the ancient human samples (Figure 2). Only one of the ten PWC individuals showed a presence of the T allele, a heterozygote, and the allele frequency for the T allele is 0.05 (1/20, with the exact 95% CI values 0.001265089 to 0.2487328, Figure 3). The T allele frequency in the PWC population differs significantly from the T allele frequency in the contemporary Swedish population (n = 97, Fisher's Exact test, p < 0.0001), where the frequency of the T allele is 0.74 (144/194, with the exact 95% CI values 0.6747198 to 0.8022533, Figure 3). Genetic drift can cause differences in allele frequencies between two samples taken from the same population separated in time. Since the PWC and extant samples are separated by a substantial amount of time (around 4000 years), a simple Fisher's exact test would not account for genetic drift, but would only be relevant for the null-hypothesis of no difference between the two samples. To explore the possibility that genetic drift alone caused the difference in T allele frequency, we can model a prehistoric population (for example the PWC population) as being ancestral to a population existing today. To evaluate different demographic scenarios (e.g. constant population sizes or population expansion from the time of the prehistoric sample), we simulated an extant sample and a prehistoric sample using the program COMPASS [17]. COMPASS simulates different temporal samples under a coalescent model, and allows a range of demographic assumptions, for example, population expansion. For a particular assumption about the demographic model, we ran a large number of replicate simulations, and kept only samples that contained exactly 144 derived alleles (the T allele) in the extant sample. In other words, we conditioned on observing exactly 144 derived alleles in the extant sample as in the empirical modern data set. Out of 1,000 replicate simulations (which all had 144 derived alleles in the extant sample), we counted the number of times zero or one derived allele was observed in the prehistoric sample. This fraction can be used to approximate the probability of observing a prehistoric sample with zero or one derived allele, conditional on observing 144 derived alleles in the extant sample. 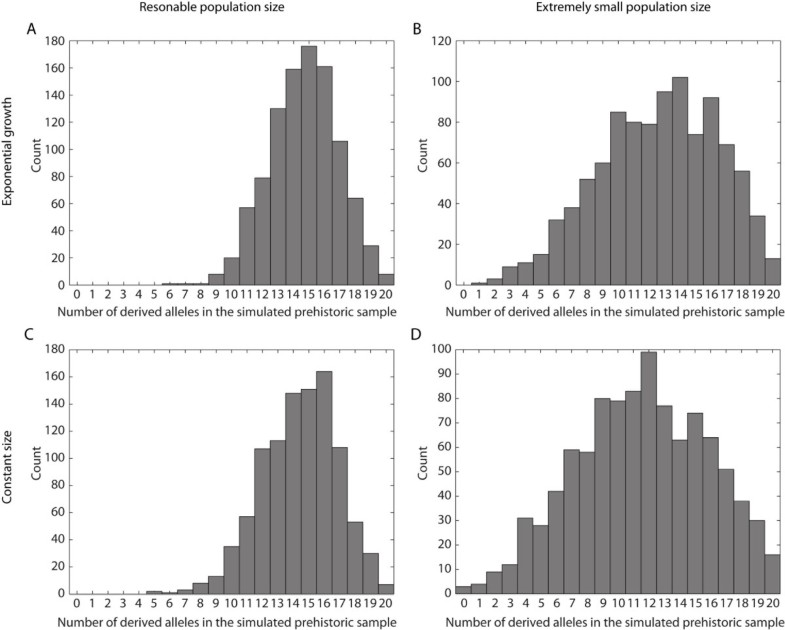 Figure 4. Simulation results for four models where one prehistoric sample and one extant sample have been collected. The distribution of a derived allele in the prehistoric sample, conditional on 144 derived alleles in the extant sample. A) The population starts to grow (exponentially) from 5,000 individuals 225 generations ago to a size of 50,000 individuals at present. B) The population starts to grow (exponentially) from 100 individuals 225 generations ago to a size of 10,000 individuals at present. C) Constant population size of 5,000 individuals. D) Constant population size of 500 individuals. Figure 4. Simulation results for four models where one prehistoric sample and one extant sample have been collected. The distribution of a derived allele in the prehistoric sample, conditional on 144 derived alleles in the extant sample. A) The population starts to grow (exponentially) from 5,000 individuals 225 generations ago to a size of 50,000 individuals at present. B) The population starts to grow (exponentially) from 100 individuals 225 generations ago to a size of 10,000 individuals at present. C) Constant population size of 5,000 individuals. D) Constant population size of 500 individuals.
Our first scenario assumes that the effective population size in Sweden is 50,000 at present and that the population grew, exponentially, from Ne = 5,000, starting 225 generations ago (before 225 generations ago, Ne was constant and 5,000). We believe that this model may be a reasonable description of the demographic history of northern Europeans based on evidence from other European populations (see e.g. [18]), but it turns out that the results are robust to relatively small effective population sizes (see below). We simulate data for one SNP under an exponential growth model, with a contemporary sample size of 194 gene-copies and a size of 20 gene-copies in the prehistoric sample. By conditioning on observing 144 derived alleles in the contemporary sample, we retain 1,000 simulated data sets, and of these 1,000 sets, no simulation had zero or one derived allele in the prehistoric sample (see Figure 4A). If we decrease the population sizes in the simulation (and thereby increase the effect of genetic drift) we can determine how robust the result is to small population sizes. Assuming that the effective population size in Sweden is 10,000 at present and that the population grew, exponentially, from Ne = 100, starting 225 generations ago (before 225 generations ago, Ne was constant and 100), we found one simulated data set (out of 1,000) that had one derived allele in the ancient sample and no simulated data set that had zero derived alleles in the prehistoric sample. Figure 4B shows the distribution of the number of derived alleles in the prehistoric sample (conditional on 144 derived alleles in the extant sample).  Discussion DiscussionAuthentication of DNA sequences in prehistoric human remains has proved complicated, mainly because modern human contamination is always present to some degree [19-21]. Here we rely on a combination of several arguments for our acceptance of the results as authentic [21-23]. The most important argument is that the success rate, like the allele frequencies, differs among the prehistoric human remains, the present-day human samples and the negative controls. Further, our material had already been pre-screened for contamination, including a major contamination investigation in which human and nonhuman DNA was typed in large parallel series of human and animal assemblages from the same sites (Figure 3) [24]. The frequency of the derived T allele, strongly associated with the ability to consume unprocessed milk at adulthood, was significantly different between the prehistoric and the extant samples. For reasonable assumptions about population sizes our simulations show that it is highly unlikely that the frequency could have shifted over time due to drift in a population with constant size, or in an expanding population. Thus, only strong positive selection or differences in population structure between the PWC population and the population ancestral to the extant Swedish population would explain the discrepancy between the extant and the prehistoric data. We note that with the data on a single SNP presented here, it is not possible to discriminate between these two scenarios. Also, it should be noted that the prehistoric samples used in this study came from a non-agricultural population, existing in parallel with an agricultural population (TRB) that is potentially ancestral to the extant Northern European population [13]. In Burger et al. [11] one Mesolithic, nine Neolithic and one Medieval samples were analyzed. In this study 9 of the 10 individuals were homozygous C at position -13.910 only the medieval individual was heterozygous for the -13.910-C/T polymorphism. These data show some similarity with our data as the Mesolithic individual does not show any lactase persistence. Here we describe a possible scenario where cultural practices could have had a tremendous impact on the genetic composition of human populations. Before dairy farming was practiced in Scandinavia, the allele frequency at -13910 was not greatly affected. The neolithisation processes in Scandinavia provided a selection pressure on this specific locus on a scale that may actually have led to a population replacement in the area (as indicated by mitochondrial DNA [13]), or at least a major shift in the allele frequency at this particular locus. This would require that cultural changes were dependent upon demographic changes in this case, and that cattle farming presented a clear advantage to hunting/gathering. In such case the 'secondary product revolution' (i.e. the introduction of milk) in northern Europe would have a large effect on the derived allele at -13910. Thus, the development that led to the present-day North European lifestyle, which is heavily based on dairy and other farm products, may have been a process where cultural practices and genes interacted. ConclusionIt is unlikely that the PWC and their ancestors used dairy products before the middle Neolithic, nor is there any archaeological evidence for such practices [25]. The difference in allele frequency from the extant Scandinavian population may therefore be explained by the fact that PWC is not ancestral to the extant Scandinavian population. An alternative, but less likely, explanation posits an ancestral relationship, but with selection dramatically increasing the frequency of the derived allele. Malmström, Helena, et al. " High frequency of lactose intolerance in a prehistoric hunter-gatherer population in northern Europe." BMC evolutionary biology 10.1 (2010): 89. |
|
|
|
Post by Admin on Sept 1, 2014 21:17:43 GMT
Idle doodle, a game of Stone Age tic-tac-toe, or the first evidence of Neanderthal art? No one can say. But two things seem reasonably clear: these scratches in the hard stone floor of a cave in Gibraltar are the work of a Neanderthal, and they were made quite purposefully more than 40,000 years ago. Some say they are abstract symbols of some description, bolstering the notion that Neanderthals were capable of subtle symbolic thought. Others remain to be convinced. The etchings were discovered by Clive Finlayson of the Gibraltar Museum and colleagues on the floor of Gorham's cave on Gibraltar's eastern shore. The cave was occupied by Neanderthals for thousands of years. The age of the thick layer of clay that lies immediately on top of the rock – itself littered with Neanderthal tools and remnants of the fires they burned – tells us that the etching was made at least 39,000 years ago.  Most experts contacted by New Scientist agreed that the scratches were made by a Neanderthal more than 39,000 years ago. Tom Higham of the University of Oxford, who recently reassessed the dates of dozens of Neanderthal sites, says the grooves must have been made before 39,000 years ago, "perhaps many millennia before". What divides opinion is what it all means. The stereotype of Neanderthals as simple, brutish early humans is now pretty much discredited. We know they hunted with knapped stone tools – and not just big game but also fish, birds and rabbits, which are thought to be trickier to catch and kill. But whether Neanderthals were capable of subtle symbolic thinking – even art – and whether they acquired this ability on their own or were inspired by the arrival of more sophisticated modern humans into Europe, has been the subject of intense debate. What does seem clear is that the etching was done with a purpose. Finlayson's colleague Francesco d'Errico of the University of Bordeaux in France carried out experiments to determine if the scratches could have been made by accident. Using two kinds of Neanderthal rock points as his stylus, and a slab of rock identical to the floor of the cave as his canvas, he needed to make in excess of 100 strokes to reproduce the pattern exactly.  Idly scratching the rock in a to-and-fro motion only scratched the surface. To get the deep, linear grooves d'Errico had to focus on the line and put his weight into the stylus. He also had to shift his body position to make the perpendicular lines. "This was not doodling," says d'Errico. "It required a lot of effort." d'Errico also tried cutting a piece of meat on the rock to see if the lines could be the by-product of butchering, but that was quickly ruled out. "Every time you cut over the meat what comes out takes a different shape," he says. "You get a scattering of lines in all directions." The only conclusion, says the team, is that the lines were deliberately made. "The pattern was clearly purposefully made, and not a utilitarian activity. There was a will to produce an abstract pattern," says d'Errico. Higham and Paul Pettitt of Durham University, UK, agree that the pattern is intentional. But while d'Errico and Finlayson claim its abstract nature says something about Neanderthal thinking, others are reluctant to take a stance. "The markings are significant if made by Neanderthals and would add to the increasing amount of information implying that they were capable of thinking in more or less abstract ways," said Higham. In a way the discovery is not even surprising. Recent discoveries have suggested that Neanderthals wore jewellery made of pierced and painted shells, and feathers, and may have buried their dead. "If the date and the species attribution stand," says April Nowell of the University of Victoria in Canada, the results "fit well with what we know about Late Neanderthal culture". |
|
|
|
Post by Admin on Sept 8, 2014 22:32:34 GMT
 At the center of the debate on the emergence of modern humans and their spread throughout the globe is the question of whether archaic Homo lineages contributed to the modern human gene pool, and more importantly, whether such contributions impacted the evolutionary adaptation of our species. A major obstacle to answering this question is that low levels of admixture with archaic lineages are not expected to leave extensive traces in the modern human gene pool because of genetic drift. Loci that have undergone strong positive selection, however, offer a unique opportunity to identify low-level admixture with archaic lineages, provided that the introgressed archaic allele has risen to high frequency under positive selection. The gene microcephalin (MCPH1) regulates brain size during development and has experienced positive selection in the lineage leading to Homo sapiens. Within modern humans, a group of closely related haplotypes at this locus, known as haplogroup D, rose from a single copy ≈37,000 years ago and swept to exceptionally high frequency (≈70% worldwide today) because of positive selection. Here, we examine the origin of haplogroup D. By using the interhaplogroup divergence test, we show that haplogroup D likely originated from a lineage separated from modern humans ≈1.1 million years ago and introgressed into humans by ≈37,000 years ago. This finding supports the possibility of admixture between modern humans and archaic Homo populations (Neanderthals being one possibility). Furthermore, it buttresses the important notion that, through such adminture, our species has benefited evolutionarily by gaining new advantageous alleles. The interhaplogroup divergence test developed here may be broadly applicable to the detection of introgression at other loci in the human genome or in genomes of other species. The extent to which anatomically modern humans admixed with archaic Homo has been the subject of repeated speculation, particularly in regards to Neanderthals (2–22). Thus far, the mainstream view from fossil and genetic studies leans toward a model where anatomically modern humans fully replaced archaic Homo lineages rather than admixed with them (2–8). However, a number of investigators have voiced opposition to this total replacement model on a number of grounds, and the debate has yet to be resolved (9–22). Particularly needed to settle this debate is the identification of genetic loci that show telltale signs of admixture. There have been several reports of loci in the human genome that display unusually deep genealogy (15, 16, 23, 24), and in some cases, admixture between humans and archaic Homo lineages has been invoked as a possible explanation. However, these studies cannot differentiate the admixture model from other possibilities, such as long-standing balancing selection, that also could contribute to deep genealogies (see Discussion). As such, proponents of the admixture scenario have yet to identify a concrete example of a genetic locus for which there is compelling evidence of admixture. Furthermore, most discussions of admixture tend to treat it as a selectively neutral event, one that happened simply as a byproduct of the geographical overlap between modern humans and archaic populations. Such discussions often overlook the possibility that admixture with archaic lineages, if it indeed occurred, might have brought adaptive alleles (along with the traits they determine) into the modern human gene pool, thus profoundly impacting the biological evolution of our species.  A major difficulty in looking for traces of ancient admixture in the modern human gene pool is that, under neutrality, very low levels of admixture are not expected to be readily detectable because of the effects of genetic drift. Indeed, simulations of ancient admixture showed that an archaic genetic contribution of <0.1% is unlikely to be detectable even in a very large set of polymorphism data (25). Thus, the absence at present of conclusive genetic data in support of the introgression scenario should not be taken as evidence against the possibility of any introgression. If introgression of archaic lineages into the modern human gene pool indeed occurred, then genes that have been subject to recent positive selection in humans may be enriched for introgressed alleles. Although selectively neutral alleles introgressed from archaic lineages at low levels are likely lost by drift or swamped by the large influx of modern human DNA, an introgressed allele that is selectively advantageous could escape the effect of genetic drift and rise to high frequency. As such, these alleles might become detectable in the modern human gene pool. The gene microcephalin is a critical regulator of brain size. In humans, loss-of-function mutations in this gene cause a condition known as primary microcephaly, which is characterized by a severe reduction in brain volume (by 3- to 4-fold) but, remarkably, a retention of overall neuroarchitecture and a lack a overt defects outside of the brain (26). The exact biochemical function of microcephalin has yet to be elucidated, but this gene likely plays an essential role in promoting the proliferation of neural progenitor cells during neurogenesis (26). microcephalin has been shown to be the target of strong positive selection in the evolutionary lineage leading from ancestral primates to humans (27, 28). This observation, coupled with the fact that this gene is a critical regulator of brain size, suggests the possibility that the molecular evolution of microcephalin may have contributed to the phenotypic evolution of the human brain (27, 28). In a recent study, we found that the haplotype structure at the human microcephalin locus is consistent with the action of recent positive selection (29). Specifically, we found that a class of haplotypes at the locus, dubbed haplogroup D, has a remarkably young coalescence age (≈37,000 years) despite an exceptionally high worldwide frequency (≈70%). This observation implies a rapid rise in the frequency of haplogroup D in humans, which is incompatible with genetic drift and instead supports the notion that positive selection has operated on haplogroup D to drive up its frequency. In the present work, we examine the origin of haplogroup D. We provide evidence that haplogroup D may have originated from a lineage separated from modern humans for ≈1.1 million years and introgressed into the human gene pool by ≈37,000 years ago. We discuss the implications of our findings for the understanding of modern human origins and the biological adaptation of our species as it spreads around the globe.  Fig. 1. Distribution of pairwise sequence divergence between and within D and non-D chromosomes at the microcephalin locus. Fig. 1. Distribution of pairwise sequence divergence between and within D and non-D chromosomes at the microcephalin locus.Highly Unusual Genealogy of the microcephalin Locus. We have examined (29) microcephalin in a panel of 89 individuals assembled to approximate the worldwide diversity of major human populations. We resequenced the panel for a 29-kb region that spans exons 4–9 of the 14-exon microcephalin gene. This process led to the identification of 220 segregating sites delineating 86 distinct haplotypes (Table 1, which is published as supporting information on the PNAS web site). Of the 178 chromosomes we sampled, 124 (or 70%) belonged to haplogroup D, defined by the derived C residue at the G37995C diagnostic nonsynonymous polymorphic site. (For simplicity, we will refer to haplogroup D as the D allele and the non-D haplotypes as the non-D allele.) Despite the high worldwide frequency of the D allele, its coalescence age is merely ≈37,000 years, far younger than the non-D allele (29). Closer scrutiny of the haplotype data in Table 1 revealed a rather peculiar pattern: the average pairwise divergence between D and non-D chromosomes across the 29-kb region is 3.3 times the divergence seen within non-D chromosomes and a striking 30 times the divergence seen within D chromosomes (Fig. 1). Further examination showed that this is because microcephalin has a highly unusual, lopsided, and deeply divided genealogy, which is schematized in Fig. 2 A.  Fig. 2. Comparison of the microcephalin genealogy with an idealized genealogy. Each filled triangle represents a genealogical clade, with the width of the triangle representing frequency in the population. (A) The genealogy consistent with the haplotype data at the microcephalin locus. The coalescence age of D chromosomes (≈37,000 years), non-D chromosomes (≈990,000 years), and between D and non-D chromosomes (≈1,700,000 years) are indicated. (B) The idealized genealogy of a partial positive selective sweep, wherein the adaptive allele first emerged by a mutational event on a random chromosome in the population. Fig. 2. Comparison of the microcephalin genealogy with an idealized genealogy. Each filled triangle represents a genealogical clade, with the width of the triangle representing frequency in the population. (A) The genealogy consistent with the haplotype data at the microcephalin locus. The coalescence age of D chromosomes (≈37,000 years), non-D chromosomes (≈990,000 years), and between D and non-D chromosomes (≈1,700,000 years) are indicated. (B) The idealized genealogy of a partial positive selective sweep, wherein the adaptive allele first emerged by a mutational event on a random chromosome in the population.Three features are prominent in this unusual genealogy. First, the D chromosomes coalesce to its most recent common ancestor (MRCA) at ≈37,000 years before present, whereas the non-D chromosomes coalesce at a far older ≈990,000 years before present. The much younger coalescence age of the D chromosomes, despite their much higher frequency, is consistent with the action of positive selection on the D allele as reported previously (29). Second, and more surprisingly, however, we found that the D and non-D chromosomes belong to two distinct, deeply divided clades connected by a single branch around the root of the tree (except for a few rare recombinants between the two clades, as discussed later). In other words, the D clade is a distant outgroup of the non-D clade, and vice versa. As shown in Fig. 3 and Table 1, the deep division between the D and non-D clades is due to a large number of segregating sites scattered throughout the 29-kb resequenced region that consistently differentiate the two clades, i.e., segregating sites for which the D chromosomes are characterized by one allele, whereas the non-D chromosomes are characterized by the other allele. For most of these segregating sites, the D chromosomes bear the derived allele, whereas the non-D chromosomes bear the ancestral allele (Fig. 3), an observation consistent with the fact that the internal branch from the MRCA of the D clade to the root of the tree is much longer than the internal branch from the MRCA of the non-D clade to the root (Fig. 2 A). Such a tree topology does not resemble the expected genealogy of a recent selective sweep, as schematized in Fig. 2 B, wherein the adaptive allele is introduced by a mutational event in a panmictic population. Fourth, whereas the coalescence age of ≈990,000 years for the non-D clade is similar to the human genome average, the D and non-D clades coalesce with each other at a much older ≈1,700,000 years before present, with virtually no genetic exchange between the two clades except for a few rare recombinants (discussed below). These unusual features of the microcephalin genealogy suggest the possibility that the MRCA of the D clade introgressed into humans from a divergent Homo lineage at or some time before ≈37,000 years ago. In the ensuing sections, we describe stringent statistical tests that support this introgression model.  Fig. 3. Distribution of congruent or near-congruent segregating sites in the 29-kb resequenced region of microcephalin. Congruent sites are defined as showing consistently different alleles between D and non-D haplotypes; near-congruent sites are defined as having no more than four differences from congruent sites. Sites for which the D chromosomes are characterized by the derived allele are indicated by long blue lines, whereas sites for which the D chromosomes are characterized by the ancestral allele are indicated by short red lines (for exact positions of these sites, see Table 1). Also indicated is the G37995C nonsynonymous site used to define the D chromosomes (bearing the derived C allele) and the non-D chromosomes (bearing the ancestral G allele).Discussion Fig. 3. Distribution of congruent or near-congruent segregating sites in the 29-kb resequenced region of microcephalin. Congruent sites are defined as showing consistently different alleles between D and non-D haplotypes; near-congruent sites are defined as having no more than four differences from congruent sites. Sites for which the D chromosomes are characterized by the derived allele are indicated by long blue lines, whereas sites for which the D chromosomes are characterized by the ancestral allele are indicated by short red lines (for exact positions of these sites, see Table 1). Also indicated is the G37995C nonsynonymous site used to define the D chromosomes (bearing the derived C allele) and the non-D chromosomes (bearing the ancestral G allele).DiscussionIn this study, we investigate the origin of the microcephalin D allele in modern humans. We show that the D allele is unlikely to have arisen within a panmictic population. Instead, our data are consistent with a model of population subdivision followed by introgression to account for the origin of the D allele. By this model, schematized in Fig. 4 B, the lineage leading to modern humans was split from another Homo lineage, and the two lineages remained in reproductive isolation for ≈1,100,000 years. During this period of reproductive isolation, the modern human lineage was fixed for the non-D allele at the microcephalin locus, whereas the other Homo lineage was fixed for the D allele. These two alleles are differentiated by a large number of sequence differences accumulated during the prolonged isolation of the two populations. At or sometime before ≈37,000 years ago, a (possibly rare) interbreeding event occurred between the two lineages, bringing a copy of the D allele into anatomically modern humans. Whereas the original D-bearing Homo population had since gone extinct, this introgressed copy of the D allele in humans had subsequently spread to exceptionally high frequency throughout much of world because of positive selection. Several studies have reported loci in the human genome that are associated with unusually deep genealogies containing highly divergent clades (15, 16, 23, 24). Although this type of observation can result from the admixture of reproductively isolated populations, the observation is itself insufficient evidence for admixture. In particular, deep divergence can occur if two or more allele classes at a locus are maintained by balancing selection for a prolonged period. So, even for a locus whose genealogy is too deep to be statistically compatible with neutral evolution, it is essential to rule out balancing selection before the admixture model can be adopted. One notable example is the MAPT locus, which has two distinct haplogroups, H1 and H2, that diverged from each other ≈3 million years ago, whereas the coalescence age of H2 is much younger (24). One explanation for this unusual observation is that H2 introgressed into modern humans from an archaic Homo lineage (17). However, because H1 and H2 are inverted relative to each other and thus cannot recombine, deep divergence between them across an extend genomic region and the young age of H2 are also compatible with balancing selection that maintained H2 at low frequencies for extended periods (along with H1) followed by a recent rise in the frequency of H2 (24). For the microcephalin locus, in contrast, we were able to rule out the possibility of balancing selection with a high degree of confidence. As such, microcephalin shows by far the most compelling evidence of admixture among the human loci examined thus far. Speculation about the identity of the archaic Homo population from which the microcephalin D allele introgressed into the modern human gene pool points to the Neanderthal lineage as a potential (although by no means only) candidate. Anatomically modern humans and Neanderthals shared a long period of coexistence, from as early as 130,000 years ago in the Middle East (39) to as late as 35,000 years ago in Europe (40), consistent with the estimated introgression time of the microcephalin D allele at or sometime before ≈37,000 years ago. Furthermore, the worldwide frequency distribution of the D allele, exceptionally high outside of Africa but low in sub-Saharan Africa (29), suggests, but does not necessitate, admixture with an archaic Eurasian population. Finally, our estimate of the separation time between D and non-D alleles (i.e., ≈1,100,000 years with a lower-bound confidence interval of ≈530,000 years) is largely consistent with the divergence time between modern humans and Neanderthals based on mitochondrial DNA (mtDNA) sequence difference (320,000–740,000 years; refs. 41 and 42) and with the earliest appearance of Neanderthals in the fossil record ≈500,000 years ago (43). It would be of great interest to sequence the microcephalin locus in Neanderthals or other archaic Homo lineages, should it become technically feasible to retrieve and analyze nuclear DNA from ancient hominid remains. Our results not only provide genetic evidence in support of the possibility of admixture between modern humans and an archaic Homo lineage but also support the notion that the biological evolution of modern humans might have benefited from the contribution of adaptive alleles from our archaic relatives. In the case of microcephalin, it is all the more intriguing given the fact that the adaptive allele is associated with an important brain development gene. As anatomically modern humans emerged from Africa and spread across the globe, the “indigenous” Homo populations they encountered had already inhabited their respective regions for long periods of time and were, in all likelihood, better adapted to the local environments than the colonizing humans, at least in some biological domains. It is perhaps not surprising then that modern humans, although likely superior in their own way, could in theory benefit from adopting some adaptive alleles from the populations they replaced. That this might indeed be the case for the brain size-determining gene microcephalin should add an important new perspective to the discussion of human origins and the recent evolution of our species. Furthermore, any admixture between modern humans and archaic populations is likely to affect more than one locus in the genome. Our study thus provides a methodological template for identifying additional loci in the human genome that might harbor alleles from archaic populations through introgression and subsequent positive selection. Evans, Patrick D., et al. " Evidence that the adaptive allele of the brain size gene microcephalin introgressed into Homo sapiens from an archaic Homo lineage." Proceedings of the National Academy of Sciences 103.48 (2006): 18178-18183. |
|

