Post by Admin on Jul 17, 2021 22:26:50 GMT
DISCUSSION
We implemented a new ARG inference approach, SARGE, and used it to build the first genome-wide ARG of both human and archaic hominin genomes. Analysis of the topology of these ARG trees confirms prior findings about archaic hominin admixture but with important new biological insights. For one, we find that a low fraction, 1.5 to 7%, of the human genome is uniquely human, with the remainder comprising lineages shared with archaic hominins from either ILS or admixture. This small human-specific fraction of the genome is enriched for genes related to neural development and function. We also find evidence for multiple waves of human-specific mutations that occurred through time, suggesting that the modern human phenotype may have developed in stages.
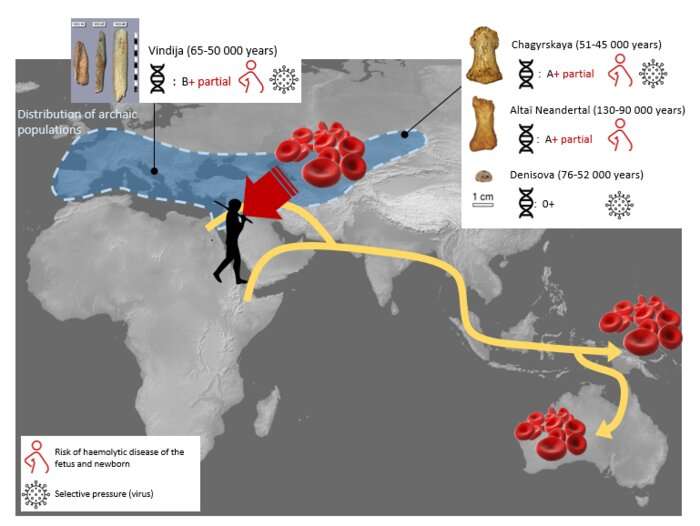
In addition to Neanderthal admixture into the ancestors of all modern non-African populations, we find evidence for other, population-specific episodes of admixture throughout Eurasia. The TMRCA to these population-specific Neanderthal haplotype blocks is deeper than the TMRCA to the Neanderthal haplotype blocks shared by all non-African populations. This deeper TMRCA suggests that Neanderthals contributing population-specific ancestry were less closely related to published (Altai and Vindija) Neanderthal genomes than were the Neanderthals that contributed the broadly shared Neanderthal haplotype blocks. We also find that Neanderthal ancestry is present to a smaller extent in some African genomes due to back-migration, consistent with other recent reports (20).
We note that our estimated TMRCA to Neanderthal within Neanderthal-introgressed segments in all non-African populations is recent, ~74 ka ago, and implies therefore that little genetic drift separates admixed humans from sequenced Neanderthals in these segments. This recent TMRCA suggests that the majority of Neanderthal ancestry in modern humans originated from Neanderthal gene flow into the ancestors of all non-Africans before populations diversified. It also suggests that at least one of the Neanderthal genomes used here is closely related to the Neanderthal(s) involved in this admixture event. The slightly elevated Neanderthal ancestry that others have described in Central and East Asian populations also appears to have originated in this first pulse, as Central and East Asian Neanderthal haplotypes are mostly shared with other, geographically distant populations. This observation favors the hypothesis that the increased Neanderthal ancestry in these populations relative to others is due to weaker selection against alleles that may be mildly deleterious (32), made possible because of smaller historical population sizes in this part of Eurasia, rather than to additional admixture events (22). Our evidence of many small-scale, population-specific admixture events, however, together with a simulation study that found a single-pulse admixture model followed by drift unable to explain the discrepancies in admixture proportions in European and Asian genomes (49), hints at a complex history of admixture throughout Eurasia not fully captured by either of these two hypotheses.
Last, the genomes of some Oceanian and other populations harbor genes from a population most closely related to the archaic Denisovan genome. The available Denisovan genome is less genetically similar to the admixing genome than the available Neanderthal genomes are to the admixing Neanderthals. While we are hopeful that future work may uncover a DNA-bearing fossil better representing the population involved in the Denisovan admixture, our approach allows identification of admixed regions that can be used to better describe the genome of the archaic hominin group involved in the admixture event. Larger panels of Denisovan admixed genomes may one day provide a nearly complete Denisovan genome scavenged in parts from the genomes of admixed human individuals.
The ARG also allows for prioritizing the selective importance of mutations specific to, and shared by, all modern humans by considering the TMRCAs of those mutations together with the lengths of their surrounding human-unique regions. Many of these selected human-specific mutations appear to affect genes involved in neural development and function, as well as RNA splicing. Using new tools for genome editing and brain organoid models for neural function, these mutations are obvious and important targets for experimental studies to determine what was selected in our human ancestors after divergence from our most closely related, extinct relatives.
Supplementary Materials
An ancestral recombination graph of human, Neanderthal, and Denisovan genomes
Nathan K. Schaefer, Beth Shapiro, Richard E. Green
Download Supplement: advances.sciencemag.org/cgi/content/full/7/29/eabc0776/DC1/1
This PDF file includes:
Supplementary Methods
Supplementary Text
Figs. S1 to S56
Tables S1 to S8
References
Files in this Data Supplement:
Adobe PDF - advances.sciencemag.org/highwire/filestream/258798/field_highwire_adjunct_files/0/abc0776_SM.pdf
We implemented a new ARG inference approach, SARGE, and used it to build the first genome-wide ARG of both human and archaic hominin genomes. Analysis of the topology of these ARG trees confirms prior findings about archaic hominin admixture but with important new biological insights. For one, we find that a low fraction, 1.5 to 7%, of the human genome is uniquely human, with the remainder comprising lineages shared with archaic hominins from either ILS or admixture. This small human-specific fraction of the genome is enriched for genes related to neural development and function. We also find evidence for multiple waves of human-specific mutations that occurred through time, suggesting that the modern human phenotype may have developed in stages.
In addition to Neanderthal admixture into the ancestors of all modern non-African populations, we find evidence for other, population-specific episodes of admixture throughout Eurasia. The TMRCA to these population-specific Neanderthal haplotype blocks is deeper than the TMRCA to the Neanderthal haplotype blocks shared by all non-African populations. This deeper TMRCA suggests that Neanderthals contributing population-specific ancestry were less closely related to published (Altai and Vindija) Neanderthal genomes than were the Neanderthals that contributed the broadly shared Neanderthal haplotype blocks. We also find that Neanderthal ancestry is present to a smaller extent in some African genomes due to back-migration, consistent with other recent reports (20).
We note that our estimated TMRCA to Neanderthal within Neanderthal-introgressed segments in all non-African populations is recent, ~74 ka ago, and implies therefore that little genetic drift separates admixed humans from sequenced Neanderthals in these segments. This recent TMRCA suggests that the majority of Neanderthal ancestry in modern humans originated from Neanderthal gene flow into the ancestors of all non-Africans before populations diversified. It also suggests that at least one of the Neanderthal genomes used here is closely related to the Neanderthal(s) involved in this admixture event. The slightly elevated Neanderthal ancestry that others have described in Central and East Asian populations also appears to have originated in this first pulse, as Central and East Asian Neanderthal haplotypes are mostly shared with other, geographically distant populations. This observation favors the hypothesis that the increased Neanderthal ancestry in these populations relative to others is due to weaker selection against alleles that may be mildly deleterious (32), made possible because of smaller historical population sizes in this part of Eurasia, rather than to additional admixture events (22). Our evidence of many small-scale, population-specific admixture events, however, together with a simulation study that found a single-pulse admixture model followed by drift unable to explain the discrepancies in admixture proportions in European and Asian genomes (49), hints at a complex history of admixture throughout Eurasia not fully captured by either of these two hypotheses.
Last, the genomes of some Oceanian and other populations harbor genes from a population most closely related to the archaic Denisovan genome. The available Denisovan genome is less genetically similar to the admixing genome than the available Neanderthal genomes are to the admixing Neanderthals. While we are hopeful that future work may uncover a DNA-bearing fossil better representing the population involved in the Denisovan admixture, our approach allows identification of admixed regions that can be used to better describe the genome of the archaic hominin group involved in the admixture event. Larger panels of Denisovan admixed genomes may one day provide a nearly complete Denisovan genome scavenged in parts from the genomes of admixed human individuals.
The ARG also allows for prioritizing the selective importance of mutations specific to, and shared by, all modern humans by considering the TMRCAs of those mutations together with the lengths of their surrounding human-unique regions. Many of these selected human-specific mutations appear to affect genes involved in neural development and function, as well as RNA splicing. Using new tools for genome editing and brain organoid models for neural function, these mutations are obvious and important targets for experimental studies to determine what was selected in our human ancestors after divergence from our most closely related, extinct relatives.
Supplementary Materials
An ancestral recombination graph of human, Neanderthal, and Denisovan genomes
Nathan K. Schaefer, Beth Shapiro, Richard E. Green
Download Supplement: advances.sciencemag.org/cgi/content/full/7/29/eabc0776/DC1/1
This PDF file includes:
Supplementary Methods
Supplementary Text
Figs. S1 to S56
Tables S1 to S8
References
Files in this Data Supplement:
Adobe PDF - advances.sciencemag.org/highwire/filestream/258798/field_highwire_adjunct_files/0/abc0776_SM.pdf