|
|
Post by Admin on May 14, 2023 19:54:43 GMT
Features of novel regions and their effects on ILDs Table 2 summarizes key features for the 22q12.1 and 1q32.3 regions discussed above as well as for the three novel (replicating) regions, most strongly associated with ILDs in the CANDELA sample. Association plots for these three regions and the associated ILDs are shown in Fig. 4. Similar information on all the other novel associated regions is presented in Supplementary Table 4 and Supplementary Note 2-3. SNPs in 3q21.1 are associated with 8 distances reflecting variation mainly in the width of the upper face with strongest association being seen with distance D213 (between landmarks 2 and 25, Figs. 1 and 4). SNPs in 8p11.21 are associated with seven distances (strongest association seen for rs59547557 with D332, involving landmarks 10 and 27, P-value 2.24 × 10−9, Fig. 4). All seven distances associated with this region are sensitive to the position of the right cheilion (Figs. 1 and 4). In previous studies, SNPs in 8p11.21 have been reported to be suggestively associated with non-syndromic cleft lip/palate43. SNPs in 10p11.1 are associated with 8 distances, with strongest association seen for rs58831446 with D511 (between landmarks 16 and 33, P-value 1 × 10−10). These 8 distances are sensitive to philtrum height (Figs. 1 and 4). Fig. 4: Regional association plots for the three novel genomic regions showing strongest association with facial features in the CANDELA sample.  Panels a–c show on the left the association P-values for SNPs in each region (index SNP has been labeled). Annotated genes in each region are shown underneath. To the right of each panel are shown the facial landmarks placed by Face++ (dark dots indicate the landmarks retained in ILD calculation). Associated ILDs are indicated with colored lines (left face: line color reflects direction and magnitude of the association; right face: line color reflects association P-value). The ILD with strongest association P-value is labeled. Genome annotation, Gene Ontology and transcription patterns in associated regions We used FUMA44 to examine genome annotations for the 186 SNPs that were significantly associated across the 33 novel regions detected in the CANDELA sample. Altogether, 91 are intergenic, 55 are intronic, 39 are ncRNA variants, and one is in a 3’ untranslated region. In line with previous analyses showing an enrichment of SNPs associated with facial features in regulatory elements active during craniofacial development4,17, we observe that SNPs in the novel regions identified here are usually near or within known craniofacial enhancers/promoters (e.g. 1q32.3 and 12q21.31, Fig. 3, Supplementary Table 4). We performed a Gene Ontology (GO) analysis for the genes nearest to the index SNPs of the novel associated regions. Consistent with previous analyses4,17, we found that these genes are significantly enriched in growth and development terms, including: GO:0006936: muscle contraction (P-value = 8.81 × 10−5), GO:0019827: stem cell population maintenance (P-value = 2.19 × 10−4), GO:0051960: regulation of nervous system development (P-value = 1.17 × 10−3), GO:0021700: developmental maturation (P-value = 2.28 × 10−3), GO:0048562: embryonic organ morphogenesis (P-value = 3.30 × 10−3), GO:0007162: negative regulation of cell adhesion (P-value = 3.83 × 10−3), and GO:0032940: secretion by cell (P-value = 8.17 × 10−3) (Supplementary Fig. 7A). To evaluate preferential transcription of genes in the newly associated regions, we contrasted publicly available RNAseq data from cranial neural crest cells (CNCC)45 to data for 318 other cell types obtained by the ENCODE project46. We found that, for the majority of the regions that could be tested (19/26), transcripts closest to the index SNPs are preferentially expressed in CNCCs, compared to other cell types, similar to what has been observed in previous analyses4,17 (Supplementary Fig. 7B). |
|
|
|
Post by Admin on May 15, 2023 19:29:54 GMT
Discussion
GWAS of facial features have identified dozens of associated genome regions1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17. In some cases, these regions overlap genes for which experimental evidence demonstrates their involvement in craniofacial development1,2,47. Furthermore, most of the SNPs associated with facial features are in non-coding regions, and enrichment analyses indicate that these SNPs are preferentially located in regulatory elements of the genome, active during craniofacial development1,2,4,17. Consistently, the novel loci (and associated SNPs) we identify here share features with findings from previous GWAS of facial morphology.
Considering the five chromosomal regions highlighted in Table 2: (i) 1q32.3 overlaps the Activating Transcription Factor 3 ATF3 gene (Fig. 3). ATF3 is an evolutionarily highly conserved transcription factor known to be involved in nervous tissue regeneration after trauma48. Although currently there is no evidence for a direct involvement of ATF3 in craniofacial development, it has been reported that ATF3 expression is regulated by FOXL2, a transcription factor whose mutations are known to lead to alterations of the midface49. Furthermore, strongest association was observed for SNPs intronic to ATF3 around an enhancer which has been shown to be active during craniofacial development50 (Fig. 3). (ii) Associated SNPs in 3q21.1 overlap the MYLK (Myosin light chain kinase) gene, which studies in mice have implicated in palate fusion during development51. (iii) The newly associated 8p11.21 region includes a cluster of disintegrin and metalloproteinase (ADAM) domain genes (Fig. 4). This is a family of surface proteins with adhesion and protease activity, members of which have been shown to be involved in craniofacial development52. Furthermore, one of the ADAM genes in the cluster on 8p11.21 (ADAM3A), has been suggestively associated with non-syndromic cleft lip/palate43. (iv) Associated SNP on 10p11.1 overlap a cluster of Zinc Finger proteins genes (ZNF, Fig. 4). This cluster includes ZNF25, which has been shown to be involved in osteoblast differentiation of human skeletal stem cells53, this is a process in which RUNX2 (a well-established craniofacial morphology gene, Table 1) also plays a major role54,55,56. (v) The mouse analyses performed here are consistent with the novel association we detect on human 22q12.1. In humans, maximum association is seen for SNPs intronic to the SEZ6L gene (Fig. 2). In mice, SNPs in Sez6l are also significantly associated, although association is strongest around the Ttc28 and Mn1 gene regions (Fig. 2). There is currently no evidence implicating SEZ6L directly in craniofacial phenotypes, but there is abundant evidence that Ttc28, Mn1 and other genes in this region are involved in mouse craniofacial development (Fig. 2)34,57,58. Interestingly, of the candidates highlighted here, three (ATF3, MYLK and SEZ6L) are the genes closest to the index SNPs and, in our RNAseq data analysis, we observe that two of these genes (MYLK and SEZ6L) are preferentially transcribed in CNCC cells (Supplementary Fig. 7B).
Genetic determinants of variation in facial features in contemporary human populations are also likely to have played a role during the evolution of facial morphology. We previously identified a region in 1p12 in which a tract introgressed from archaic humans (Denisovans) impacts on lip thickness. That chromosomal region had previously been shown to be associated with body fat distribution59 and bears a strong signature of natural selection, raising the possibility that Denisovan introgression could have facilitated adaptation to a cold environment. The evidence we observe of Neanderthal introgression in 1q32.3 impacting on mid-face height represents the second instance of archaic human introgression affecting facial morphology in modern humans. In this case, the possibility of examining similar skull traits in contemporary human and Neanderthal skulls allowed us to determine that the increase in mid-face height associated with archaic introgression in 1q32.3 is consistent with the modern human-Neanderthal morphological differentiation. Evaluating the consistency of phenotypic effects had not been possible in the case of Denisovan introgression in 1p12 as that case concerned only soft tissues (the lips). Analysis of skulls has long shown that facial morphology differs markedly between Neanderthals and modern humans with the mid-face, particularly the nasal cavity, showing major differences60. This includes markedly taller noses in Neanderthals than in modern humans. Furthermore, it has long been speculated that nose morphology (in Neanderthals as well as in modern humans) has been the subject of natural selection, particularly as an adaptation to environmental temperature and humidity61,62,63. Further genetic work, including future analyses of additional ancient DNA samples, could help shed light on this question.
Although the earliest (and largest) studies on the genetics of facial variation have been carried out in people of Europeans ancestry4,9,10, recent efforts have increasingly sought to examine non-Europeans1,2,5,32. Populations with admixed continental ancestry, such as Latin Americans, offer challenges and opportunities for such studies. In these populations, optimal correction for population stratification, considering both global and local genomic ancestry, is a challenging analytical problem for which an all-round solution is yet to be developed64,65,66. Use of genetic PCs and local ancestry correction approaches to deal with population stratification should therefore be undertaken with caution. Nevertheless, the extensive genetic and phenotypic diversity of Latin Americans is enabling GWASs that have led to important insights into the genetics of physical appearance1,2,22,23,24. This is illustrated here by the novel instance of archaic introgression detected in 1q32.3: the introgressed tract has a high frequency in Native Americans but is essentially absent in Europeans (Table 2, Supplementary Fig. 6). Given the wide-spread availability of 2D photographs, the automated landmarking approach we applied here could facilitate a more comprehensive world-wide sampling of human facial variation than hitherto attempted. The study of larger and more diverse study samples should enable a fuller assessment of the genetic architecture of facial variation in the global human population and of the evolutionary forces that have shaped this variation across the world.
|
|
|
|
Post by Admin on May 15, 2023 22:23:51 GMT
A new analysis of Neanderthal genomes suggests the ancient blood that once pumped through this long-extinct population of archaic humans had more in common with modern human blood than scientists realized. While it was long assumed that Neanderthals all possessed blood type O, a new study of previously sequenced genomes of three Neanderthal individuals shows polymorphic variations in their blood, indicating they also carried other blood types found in the ABO blood group system. This means Neanderthal blood not only came in the form of blood type O – which was the only confirmed kind before this, based on a prior analysis of one individual – but also blood types A and B. The finding, based on a range of blood group alleles identified in the genetic sequences of the three Neanderthal individuals (and also one Denisovan), appears to have been hiding in plain sight, with the team who made the discovery suggesting blood group research like this has been neglected amidst the emergence of other DNA analysis technologies. 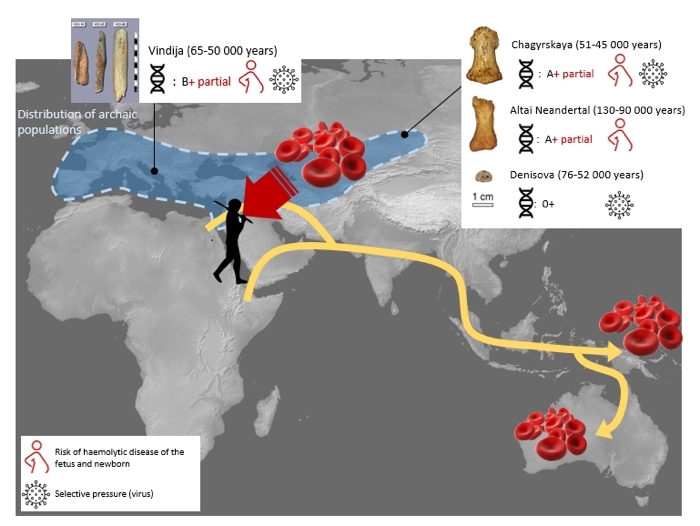 "Red cell blood groups are powerful anthropological markers," the researchers, led by first author and paleoanthropologist Silvana Condemi from Aix-Marseille University in France, explain in a new paper. "Curiously, despite their significance and the amount of available genotypic data on modern humans that is continuously accumulating, almost no attention has been paid to … major red cell blood polymorphisms in paleogenetic studies." In addition to the ABO discoveries, the researchers say the genes underlying the blood groups in these archaic humans consolidate the view that Neanderthals and Denisovans originally emerged out of Africa – due to an absence of certain antigens in their blood and the presence of ancestral blood groups linked to African populations. "These features are in accordance with a Neanderthal and Denisovan gene pool pre-dating the exit of Homo sapiens from Africa," the researchers write. Other genetic clues speak to a much later passage of time, including an allele of the RHD gene (which codes a protein in the Rh blood group), that's not found in modern humans with the exception of two present-day individuals: an Aboriginal Australian and an indigenous Papuan. It's likely, the researchers suggest, that this mysterious link is evidence of interbreeding between Neanderthals and H. sapiens before the latter migrated to Southeast Asia. As for what later helped spur the decline of the Neanderthals, the new study offers some ideas on that front too. According to the researchers, a large number of shared alleles seen in the archaic genomes of the three Neanderthals and the Denisovan suggests low genetic diversity, likely linked to inbreeding in the case of the Neanderthals. Furthermore, genetic variants in the archaic blood borne by the Neanderthals would have made them much more likely to develop hemolytic disease of the newborn (HDFN), an alloimmune condition in which a mother's immune system attacks the blood cells of her unborn fetus. "These elements could have contributed to weakening the descendants to the point of leading to their demise especially combined with the competition with H. sapiens for the same ecological niche," the researchers write. The findings are reported in PLOS One. journals.plos.org/plosone/article?id=10.1371/journal.pone.0254175 |
|
|
|
Post by Admin on May 15, 2023 22:33:48 GMT
The authors took advantage of open access to drill down into the high-quality genomes of three Neanderthals and a Denisovan – for some reason all of them female. The Neanderthals included two Siberians: the 100,000 year old Altai female who lived in Denisova Cave, and a 48,000 year female who resided in Chagyrskaya Cave. A third female aged about 57,000 years came from Vindija Cave in Croatia. The Denisovan genome came from a female who lived in Denisova Cave about 64,000 years ago. The first surprise was discovering that the full variability of the ABO system seen in modern humans was present in the Neanderthals. “We thought for years that H. sapiens was the only one to have the full set,” says Condemi. That was because our closest relatives have only a partial set. Chimpanzees are all type A; gorillas are all type B. Until this study the only Neanderthal to be checked was blood type O. “So the assumption was they were all O,” says Condemi. In fact the Vindija lady was a B (genotype BO), the Chagyrskaya lady an A (genotype AA), and the Altai lady A (genotype AA). The most parsimonious explanation, says Condemi, is that the African ancestor of Neanderthals and modern humans, perhaps Homo heidelbergensis, already possessed the full range of the ABO system. The next surprise was that all three Neanderthals carried a rare Rhesus type which Condemi refers to as “Rhesus plus incomplete”. This variant had only ever been seen once before. In 2019, researchers analysing the DNA of 72 Western Desert Aboriginal people found that one of them carried the same novel Rhesus type. onlinelibrary.wiley.com/doi/10.1111/trf.15047“At the time, it was assumed to be a new Rhesus type that had arisen in Australia,” says Condemi. “Now we know that it had existed in the past and was lost.” The authors also found that in a population of 80 people from Papua New Guinea, only one carried this rare Rh type. science.sciencemag.org/content/367/6484/eaay5012?rss=1The finding confirms the evidence from DNA (all non-Africans carry about 2% Neanderthal DNA) that modern humans interbred with Neanderthals in the Middle East before heading to south Asia and Australia. The blood-type findings also gives clues as to the Neanderthals’ disappearance. First, there’s the finding that three Neanderthals separated by 50,000 years in time and 5,000 km of space all shared the same Rhesus type. This adds to the evidence from genome studies of their low genetic diversity, a factor that can put a species at risk of being wiped out by disease. On the other hand, interbreeding with modern humans could have put them at risk of another kind. Condemi says that if an Rh ‘partial complete’ Neanderthal mated with an Rh complete H. sapiens, there would be an 18% chance of the infant developing the condition known as “haemolytic disease of the newborn” and dying. Bottom line: if you need to give a Neanderthal a blood transfusion, don’t use human blood unless it’s from a rare Western Desert Australian or Papuan. |
|
|
|
Post by Admin on May 16, 2023 2:07:07 GMT
Blood groups of Neandertals and Denisova decrypted Abstract Blood group systems were the first phenotypic markers used in anthropology to decipher the origin of populations, their migratory movements, and their admixture. The recent emergence of new technologies based on the decoding of nucleic acids from an individual’s entire genome has relegated them to their primary application, blood transfusion. Thus, despite the finer mapping of the modern human genome in relation to Neanderthal and Denisova populations, little is known about red cell blood groups in these archaic populations. Here we analyze the available high-quality sequences of three Neanderthals and one Denisovan individuals for 7 blood group systems that are used today in transfusion (ABO including H/Se, Rh (Rhesus), Kell, Duffy, Kidd, MNS, Diego). We show that Neanderthal and Denisova were polymorphic for ABO and shared blood group alleles recurrent in modern Sub-Saharan populations. Furthermore, we found ABO-related alleles currently preventing from viral gut infection and Neanderthal RHD and RHCE alleles nowadays associated with a high risk of hemolytic disease of the fetus and newborn. Such a common blood group pattern across time and space is coherent with a Neanderthal population of low genetic diversity exposed to low reproductive success and with their inevitable demise. Lastly, we connect a Neanderthal RHD allele to two present-day Aboriginal Australian and Papuan, suggesting that a segment of archaic genome was introgressed in this gene in non-Eurasian populations. While contributing to both the origin and late evolutionary history of Neanderthal and Denisova, our results further illustrate that blood group systems are a relevant piece of the puzzle helping to decipher it. Introduction Over the last decade, technological progress has allowed generation of data from the entire genome of some fifteen extinct Neanderthal and Denisova hominins who lived 40,000 to 100,000 years ago from Western Europe to Siberia [1]. It reveals population structure, several demographic fluctuations and gene flows across hominin populations, worldwide dispersal of archaic genes by admixed Homo sapiens, and even the existence of a super-archaic ‘ghost’ population [2]. In addition, the availability of Neanderthal and Denisova DNA sequences provides a phylogenetic status and chronological depth that have significantly enhanced the understanding of gene variation in modern humans for phenotype, metabolic, and immune traits (reviewed in [3]). Red cell blood groups are powerful anthropological markers. Phenotype and genotype geographical distribution mirrors past human migrations and natural selection [4–7] and comparison with primates makes it possible to evoke their evolutionary and migration trajectory with accuracy [8, 9]. Red cell blood groups are also crucial in medicine to ensure transfusion safety, transplants, and foeto-maternal compatibility [10]. To date, the International Society of Blood Transfusion (retrieved from the ISBT website, has recorded more than 380 blood group specificities grouped into 40 systems. In transfusion, it is routine practice to scrutinize six blood groups: ABO, Rh, Kell, Duffy, Kidd and MNS (reviewed in [10]). Curiously, despite their significance and the amount of available genotypic data on modern Humans that is continuously accumulating [11], almost no attention has been paid to these major red cell blood polymorphisms in palaeogenetic studies [12]. In the present study, we analyze Neanderthal and Denisovan blood groups in order to trace back the current human diversity and to discuss health aspects and vulnerabilities of archaic populations. For that purpose, we investigated the high-quality nuclear genomes previously published from three Neanderthals one Denisovan. |
|

