|
|
Post by Admin on Jul 6, 2022 19:43:36 GMT
The rs1773054 enhancer interacts with the LZTFL1 promoter The 3p21.31 locus is gene-dense and contains several candidates that could potentially be involved in COVID-19 pathogenesis. These include three chemokine receptors: CCR9 (which encodes a lymphocyte-expressed C-C chemokine receptor41); CXCR6 (which is associated with sarcoidosis and is a coreceptor for HIV42,43); and XCR1 (which encodes a X-C chemokine receptor). Transcriptome-wide association study (TWAS) analysis also identified CCR2, CCR3 and FYCO1, which lie up to 500 kilobases (kb) away, as candidate effector genes for the 3p21.31 COVID-19 association10. In addition, there are the two nearest genes that are less well studied: SLC6A20 (the SIT1 imino acid transporter associated with glycinuria44) and LZTFL1 (ref. 45), the homozygous loss of which causes the classical ciliopathy Bardet–Biedl syndrome46,47. To identify candidate target genes of the rs17713054 enhancer we performed NuTi Capture-C20,21 from the promoters of genes in surrounding regulatory domains (Methods) in primary human umbilical vein endothelial cells (HUVECs) where the rs17713054 enhancer is accessible, as well as resting and stimulated primary CD4+ T cells, primary CD14+ monocytes, CD71+ CD235+ erythroid cells and H1 human embryonic stem cells (H1-hESCs), where the enhancer is not accessible. In all cell types tested, all 28 COVID-19-associated variants fell within a domain of interaction that contained only the promoters of LZTFL1, SLC6A20 and CCR9, and is delimited by convergent CTCF boundary motifs (Fig. 3a). Within this domain, the promoters of both LZTFL1 and SLC6A20 interacted more strongly with the rs17713054 enhancer than CCR9 (Fig. 3b). Reciprocal Capture-C from the rs17713054 enhancer also showed that its interactions were primarily constrained to the same domain (Extended Data Fig. 7a). Notably, inside this domain, several tissue-specific enhancers could be seen for immune, erythroid and endothelial cell types, altering the interaction profile of the ubiquitously accessible LZTFL1 promoter and indicating dynamic regulation (Supplementary Fig. 1). Fig. 3: The interaction landscape of the severe COVID-19 risk locus. 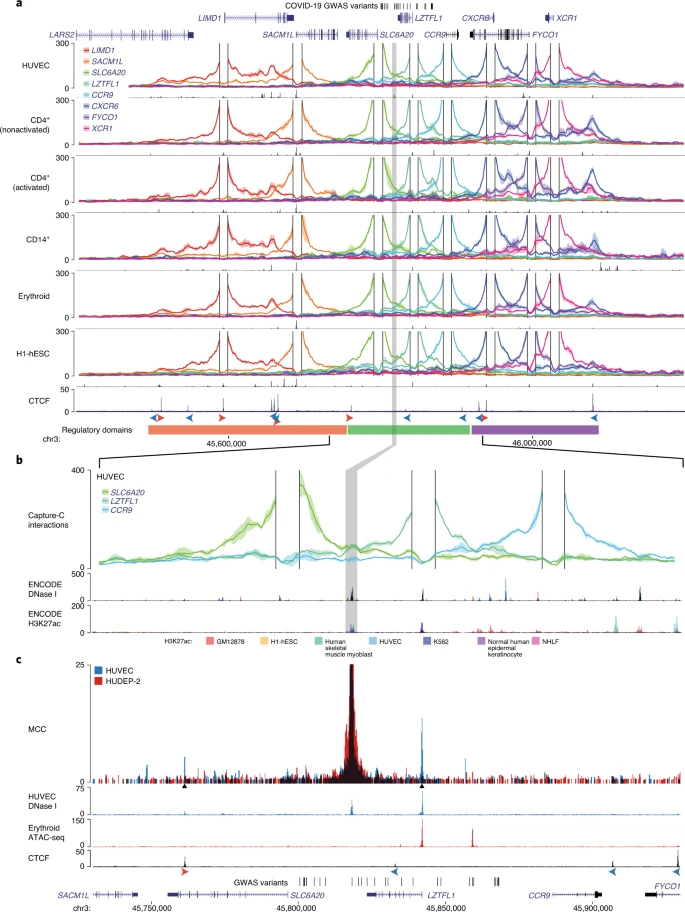 a, DpnII Capture-C-derived mean interaction count (n = 3 for all except CD14+, n = 2) and 1 s.d. (shading) for gene promoters in HUVECs, resting and activated T cells (CD4+ nonactivated/activated), monocytes (CD14+), CD71+ CD235+ erythroid cells and H1-hESCs. The enhancer containing rs17713054 is highlighted by a gray box. ATAC-seq/DNase I for each cell type is shown underneath in black. The CTCF track shows binding of the CCAAT-binding factor that acts as a boundary with forward and reverse motif orientation shown with arrowheads (red and blue, respectively). Three broad regulatory domains were identified as regions with overlapping interactions (region: chr3:45,400,000–46,200,000, hg38). Per-fragment interactions were smoothed using 400-bp bins and an 8-kb window. b, The rs17713054 regulatory domain in endothelial cells (HUVECs). Overlaid DNase I shows accessible sites in 95 cell types and H3K27ac shows active elements (region: chr3:45,730,000–45,930,000, hg38). Per-fragment interactions were smoothed using 250-bp bins and a 5-kb window. The solid line shows the mean interaction count (n = 3 independent samples) with 1 s.d. (shading). c, MCC of the rs17713054 enhancer in endothelial (HUVECs, blue) and erythroid (HUDEP-2, red) cells with tissue-specific open chromatin tracks (n = 3). Peak analysis of MCC using LanceOtron to compare the HUVEC and HUDEP-2 profiles identified two significantly enriched peaks in HUVEC cells (black triangles, P ≤ 1 × 10−999) that correspond to the LZTFL1 promoter and upstream CTCF site. We went on to perform Micro Capture-C (MCC), a 3C method that provides higher resolution data than conventional approaches22, from the rs17713054 enhancer in endothelial cells. MCC in HUVECs delineated significant tissue-specific interaction with the LZTFL1 promoter and the nearest upstream boundary CTCF site but no other significant peaks of interactions with any of the other gene promoters in the region (Fig. 3c and Extended Data Fig. 7a). Importantly, we did not find a peak of interaction with SLC6A20, probably because ENCODE datasets show that SLC6A20 carries Polycomb repression marks in endothelial (HUVEC) and normal human lung fibroblast (NHLF) cells (Extended Data Fig. 7b). Additionally, the LZTFL1 promoter was more consistently accessible in cells where rs17713054 was also accessible (Extended Data Fig. 7c,d). Therefore, LZTFL1 is the most likely direct regulatory target of the rs17713054-containing epithelial–endothelial–fibroblast enhancer. |
|
|
|
Post by Admin on Jul 7, 2022 16:17:31 GMT
rs17713054 A is associated with higher gene expression in the lung Disease biology, deepHaem, TWAS analysis10 and a phenome-wide association study11 identified lung tissue and function as key for the 3p21.31 COVID-19 association. Analysis of whole-lung RNA-seq28 showed that LZTFL1 is strongly expressed in the lung (Fig. 4a) and single-cell RNA-seq (scRNA-seq)48 showed that LZTFL1 is present throughout the respiratory epithelium but predominantly expressed in ciliated cells (Fig. 4b,c). Of the other candidate genes identified in this study and elsewhere10,49,50 (SLC6A20, CCR2, CCR3, CCR9, CXCR6 and FYCO1), only SLC6A20 and FYCO1 were consistently expressed in both lung bulk and scRNA-seq datasets, although CCR2 and CXCR6 were found in bulk RNA-seq. FYCO1 was found in most cell types and SLC6A20 was restricted to goblet cells and alveolar type 2 pneumocytes (Fig. 4 and Extended Data Fig. 8). Analysis using the Genotype-Tissue Expression28 (GTEx) portal for expression quantitative trait loci (eQTLs) showed that the rs17713054 A risk allele was associated with higher levels of expression in the lung of LZTFL1 and SLC6A20 but not the other genes (Fig. 4d and Extended Data Fig. 8). Colocalization analysis51 showed that these GWAS and eQTL associations are more likely as a result of a single variant (posterior probability (PP) = 0.2657) than two distinct variants (PP = 0.0566). Fig. 4: Pulmonary expression analysis of LZTFL1 and SLC6A20. 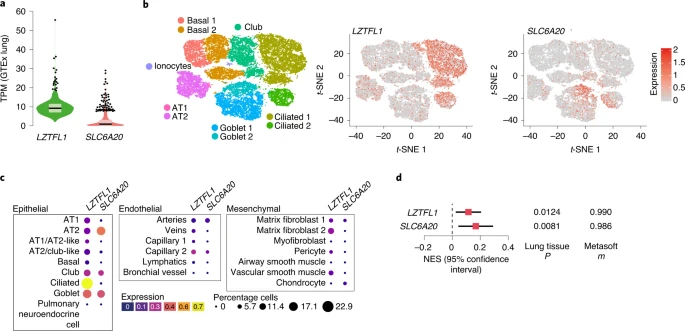 a, GTEx whole-lung RNA-seq expression profiles for LZTFL1 and SLC6A20 as transcripts per million (TPM). For the violin plots, minima and maxima are the top and bottom of the violin, the black lines show the means, the ends of the pale regions denote the first and third quartiles and the black dots denote outliers (n = 578 independent samples). b, 10x Genomics Chromium droplet scRNA-seq from the upper and lower airways and lung parenchyma48 from healthy volunteers or deceased transplant donors with ten epithelial populations (left). scRNA-seq expression profiles for LZTFL1 (middle) and SLC6A20 (right). c, Chromium single-nucleus RNA-seq35 from non-diseased adult lung (n = 3) with 22 epithelial, endothelial and mesenchymal populations, including AT1 and AT2 pneumocytes. d, GTEx eQTL analysis the rs17713054 risk A allele in the lung (n = 515 independent samples). The normalized effect size (NES) is the slope of the linear regression comparing the alternate (A) allele to the reference (G) allele. NES are calculated in a normalized space where magnitude has no direct biological interpretation. The lines show the 95% confidence interval, with significance values for single-tissue (two-sided P value without multiple test correction) and multi-tissue (PP/m value) analyses. CRISPR–Cas9 genome editing52 allows the possibility to test the role of the rs17713054 enhancer in the regulation of LZTFL1 and SLC6A20. Since the enhancer shows accessibility in epithelial, endothelial and mesenchymal cells (Extended Data Fig. 9a), we used CRISPR–Cas9 ribonucleoprotein (RNP) editing to delete either a 108- or 191-bp region at high efficiency (>70%) from H441 distal lung epithelial cells, adult blood outgrowth endothelial cells, HUVECs and IMR-90 lung fibroblast cells (Extended Data Fig. 9b–d and Supplementary Fig. 2). Using real-time quantitative PCR (qPCR) we detected no effect on LZTFL1 expression after enhancer deletion (Extended Data Fig. 9e), which is consistent with a study that CRISPR interference in the 16HBE14o- bronchial epithelial cell line had no effect on nearby gene expression50. Since SLC6A20 is Polycomb-repressed in fibroblasts and endothelial cells, it was undetectable by qPCR with reverse transcription (RT–qPCR). To understand the unexpected result, we generated H3K27ac ChIP-seq in all four cell types (Extended Data Fig. 9f,g). The rs17713054 enhancer lacked strong H3K27ac and was probably inactive, explaining the lack of effect seen by deletion. Therefore, a suitable cell model for testing the effects of rs17713054, particularly in the lung epithelium, is not currently available. |
|
|
|
Post by Admin on Jul 8, 2022 1:00:48 GMT
Epithelial dysfunction in the COVID-19 lung Given that the rs17713054 enhancer is present and LZTFL1 is expressed in lung epithelial cells, the respiratory epithelium is of particular interest for understanding the association at 3p21.31. EMT, a developmental pathway that allows terminally differentiated epithelial cells to dedifferentiate and acquire mesenchymal identity, plays a key role in the innate immune response, is a consequence of lung inflammation and is involved in both the development and resolution of pneumonitis53,54,55,56. SARS-CoV-2 is known to induce EMT in both lung carcinoma cell lines and in the respiratory tract57,58 and LZTFL1 is known to regulate EMT through Wnt/β-catenin, hedgehog and transforming growth factor-β (TGF-β) signaling59,60. In the context of malignancy, increased levels of LZTFL1 inhibits EMT, whereas decreased LZTFL1 promotes EMT45,59,60. Defining EMT in complex tissues is challenging due to its diverse and dynamic nature but can be achieved through a combined assessment of cellular reorganization, an abundance of fibroblasts (which are a product of EMT), presence of EMT-promoting signaling pathways and coexpression of epithelial and mesenchymal markers61. Consistent with the work by others62,63, we saw widespread epithelial dysfunction and diffuse alveolar damage with reorganization indicative of EMT evident in postmortem biopsies of three patients with COVID-19. Dysfunction in ciliated airways included denudation, hyperplasia and squamous metaplasia (Fig. 5a). Features of diffuse alveolar damage included pneumocyte hyperplasia, hyaline membrane deposition, immune inflammation, fine and focal fibrosis and squamous metaplasia (Fig. 5b). Between the areas of interstitial expansion and fibrotic foci, there was an accumulation of fibroblasts, which is generally absent from healthy lung tissue. Fig. 5: The lungs of patients with COVID-19 show signals of EMT. 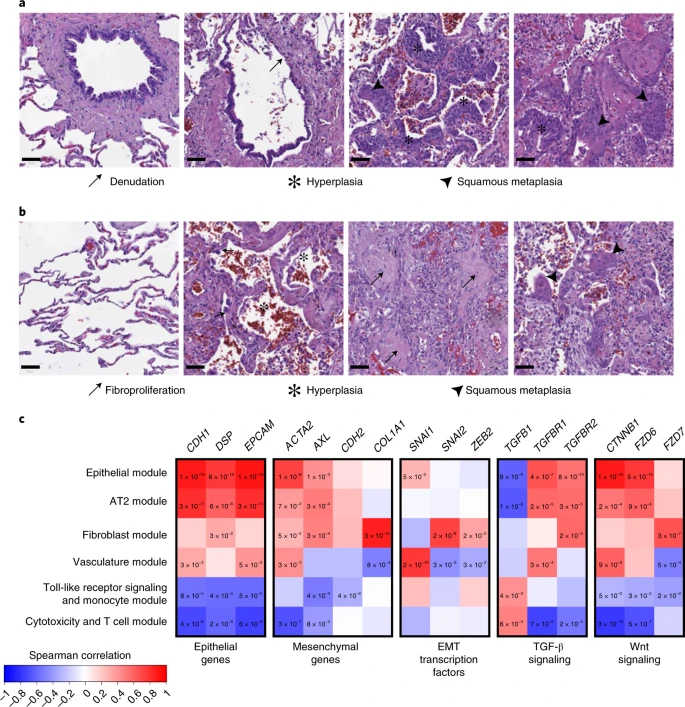 a,b, H&E-stained biopsies of the ciliated respiratory epithelium on bronchiole (a) and of alveolar space (b) in healthy lung (left) and the lung of patients with COVID-19 (middle and right). The samples of patients with COVID-19 are representative images from the staining of biopsies from three individuals and show loss of ciliated cell-lined bronchioles (denudation) and loss of alveolar monolayers populated by AT1 pneumocytes with few AT2 pneumocytes, with alveolar wall expansion and fine interstitial fibrosis. Scale bars, 50 µM. c, Spearman correlation of gene-expression profiles for EMT-related genes with the eigengenes of cell type modules identified by WGCNA analysis from spatially resolved expression data from the lung of patients with COVID-19. P values were identified by two-sided Hmisc analysis (without multiple test correction); values for significant correlations (P < 0.05) are shown and all correlation and P values are in the Source data. We previously generated selective spatial transcriptomics from 46 areas of postmortem biopsies from patients with critical COVID-19 covering a spectrum of alveolar injury64. To explore the expression profiles of EMT-relevant genes we used both a cell deconvolution approach65, to estimate cell abundance through gene transcripts, and a weighted gene correlation network analysis66 (WGCNA), to identify modules of coregulated gene-expression patterns that were assigned to cell types or biological processes. As expected, epithelial marker genes (CDH1, EPCAM) were naturally associated with alveolar type (AT) 1 and AT2 pneumocytes, as well as both of the epithelial and AT2 pneumocyte WGCNA modules (Fig. 5c and Extended Data Fig. 10). However, AT1 was also positively associated with the hallmark EMT gene ACTA2 (actin alpha 2, smooth muscle; Hmisc rcorr asymptomatic P = 0.0014), as were both the AT2 and epithelial modules (P = 0.0069 and P = 9.59 × 10−9, respectively). These two modules were also positively associated with a second mesenchymal EMT marker gene, the receptor tyrosine kinase encoding AXL (P = 0.0002 and P = 0.0031). We next investigated EMT-associated transcription factors, finding SNAI1 (snail family transcriptional repressor 1) positively associated with the epithelial module (P = 0.0491) and AT1 cells (P = 0.0432), while fibroblasts were associated with SNAI2 (P = 1.08 × 10−6) and the fibroblast module was associated with both SNAI2 (P = 1.54 × 10−8) and ZEB2 (zinc finger E-box binding homeobox 2; P = 0.0144). Finally, we investigated the Wnt/β-catenin and TGF-β pathways, finding that both pneumocyte subtypes (AT1, AT2) and both epithelial modules were associated with TGF-β signaling receptor genes (TGFBR1 and TGFBR2) and Wnt signaling genes that encode β-catenin and frizzled receptors (CTNNB1 and FZD6). By contrast, neither CD8+ T cells nor the cytotoxicity and T cell module expressed epithelial or mesenchymal genes but they expressed TGFB1 (P = 0.0029 and P = 0.0005, respectively). The colocalized expression of mesenchymal genes with epithelial cells, along with the expression of EMT transcription factors and associated signaling pathways is indicative of the EMT process, highlighting the relevance of this cellular reorganization pathway in COVID-19. Therefore, the modulation of EMT by LZTFL1 may be of relevance to the pathological outcome of COVID-19 infection. |
|
|
|
Post by Admin on Jul 8, 2022 13:31:32 GMT
Discussion
We applied a machine learning and molecular biology platform for decoding GWAS hits and identified a relatively unstudied gene, LZTFL1, as a candidate causal gene potentially responsible for the twofold increased risk of respiratory failure from COVID-19 associated with 3p21.31. The risk allele of the SNP, rs17713054 A, leads to increased transcription through augmentation of an epithelial–endothelial–fibroblast enhancer, facilitated by the addition of a second CEBPB binding motif.
MCC identified LZTFL1 as the only gene to specifically interact with the rs17713054 enhancer. However, it is possible LZTFL1 may not be the sole causal gene at 3p21.31. Two TWAS identified 11 candidate genes at this locus10,49, including LZTFL1 and SLC6A20, but only these two genes have strong 3C contacts with the rs17713054 enhancer and lung eQTLs. TWAS cannot differentiate between direct and indirect regulation67. The absence of a 3C interaction with COVID-19 severity-associated variants suggests that there may be an indirect effect for other genes, with the caveat that it is possible that a direct effect may occur in an untested cell type. While the ultrahigh resolution MCC approach only identified physical contacts between LZTFL1 and rs17713054, traditional 3C found both CCR9 and SLC6A20 to be in the same regulatory domain. CCR9 is not expressed in the lung and rs17713054 is not in an active enhancer in immune cells, where CCR9 is expressed. Both LZTFL1 and SLC6A20 have higher expression in the presence of the rs17713054 risk allele; it is plausible that in cells where SLC6A20 is not Polycomb-repressed (for example, goblet cells and AT2 pneumocytes), it also directly interacts with the rs17713054 enhancer and would thus be affected by the risk allele.
The biological relevance of SLC6A20 to COVID-19 is unclear. It is primarily expressed in the kidneys and gastrointestinal tract and its associated Mendelian disease causes renal calculi due to failure of reuptake of glycine in the nephron44. Nevertheless, its function as an imino acid transporter is modulated by levels of angiotensin-converting enzyme 2 (ref. 68) (ACE2), which is a cell receptor for SARS-CoV-2 (ref. 69). Conversely, LZTFL1 is widely expressed in pulmonary epithelial cells, including ciliated epithelial cells, which have been identified as one of the main cellular targets for SARS-CoV-2 infection70. Furthermore, homozygous loss of LZTFL1 causes a classical ciliopathy––Bardet–Biedl syndrome46,47. The association of 3p21.31 variants with susceptibility to SARS-CoV-2 infection, as well as disease severity, highlights the importance of the respiratory epithelium for this locus11. LZTFL1 encodes a cytosolic leucine zipper protein, which associates with the epithelial marker E-cadherin and is involved in the trafficking of numerous signaling molecules45,71,72,73,74. We note that upregulation of LZTFL1 in the context of malignancy inhibits EMT45,59,60, a pathway known to be part of both wound healing and immune responses53,54,55,56.
Examination of postmortem COVID-19 lung biopsies demonstrated widespread epithelial dysfunction with EMT signatures62,63. Consistently, scRNA-seq showed a reduction in the total numbers of epithelial cells after infection75, with a lower epithelial composition correlating with a more rapid progression from symptom onset to death76. The samples analyzed in this study showed few areas of healthy tissue and it is possible that inflammation or neutrophil extracellular traps, rather than direct viral infection, was driving this epithelial dysfunction58 and that LZTFL1 acts earlier in disease progression, contributing to poor structural resolution of inflammation. Expression profiling of nasal epithelia from patients with COVID-19 detected EMT signals in the upper respiratory tract57. Similarly, SARS-CoV-2 infection of both a reconstructed human bronchial epithelium model and Syrian hamster induced dedifferentiation of airway ciliated cells77, highlighting the relevance of this pathway and cell type. As such, an effect of the 3p21.31 locus in the early epithelial response may contribute to susceptibility to SARS-CoV-2 infection11. Although both influenza and SARS-CoV-2 have been shown to induce EMT57,78, its role in viral infection is not entirely clear. While chronic EMT leads to fibrosis and severe inflammation, acute EMT may be a beneficial response. In the context of viral infection, EMT leads to a reduction of two of the cell receptors of SARS-CoV-2: ACE2 and transmembrane protease serine 2 (TMPRSS2) (refs. 57,79). A reduction in these cell surface markers as a result of EMT could reduce viral load by decreasing infection efficiency and preventing severe disease. Conversely, EMT allows for epithelial cells to proliferate, repair damaged tissue and replace lost cells, which may be required to overcome severe disease.
For the 3p21.31 COVID-19 risk locus, higher risk is associated with increased expression of LZTFL1, a known EMT inhibitor. Higher levels of LZTFL1 may delay the positive effects of an acute EMT response, blocking a reduction in ACE2 and TMPRSS2 levels and/or through slowing EMT-driven tissue repair. Further investigation of the potential role of LZTFL1 and EMT in pulmonary pathogenesis is needed. Our findings suggest that a gain-of-function variant in an inducible enhancer, causing increased expression of LZTFL1, may be associated with a worse outcome. This raises the possibility that LZTFL1 could be a potential therapeutic target for the treatment or prevention of COVID-19.
|
|
|
|
Post by Admin on Jun 19, 2023 20:04:38 GMT
Neanderthal DNA constitutes up to 4 percent of the modern human genome, but the exact impacts of this DNA have remained largely unknown—until now. A team of researchers from across the United States came together to assess how these ancient genes affect modern humans, compiling their results in a paper published in the journal eLife in March. Specifically, they were able to pinpoint the involvement of Neanderthal DNA in human development, metabolism and the immune system. "The overall effect of Neanderthal alleles on metabolism/immune system is complex," Sriram Sankararaman, a co-author on the study and Professor of Computer Science, Genetics and Computational Medicine at the University of California, Los Angeles, told Newsweek. "In our study, we were able to pinpoint the impact of specific introgressed Neanderthal [gene variants]. For example, we were able to find instances of introgressed [gene variants] that impact the function of genes known to be important for immune function."  Neanderthal female One of the most significant examples of this was a genetic variant that affects the structure of an important receptor on the surface of white blood cells. This receptor is involved in the process of phagocytosis—when white blood cells engulf bacteria, parasites and infected cells. The Neanderthal variant of this gene would therefore likely disrupt our cells' ability to clear infections, making us more prone to getting sick. A different Neanderthal variant was found to disrupt a key component of cellular energy production. This builds on previous studies reporting that Neanderthal DNA may be an important contributor to COVID-19 susceptibility. Having Neanderthal DNA isn't necessarily bad, though. "A number of studies have shown that Neanderthal DNA can have a beneficial impact," Sankararaman said. "The most striking example is a region in a gene BNC2 associated with skin and hair color, where more than half of present-day Europeans carry Neanderthal DNA likely due to the adaptive benefit conferred by this introgressed DNA. "There are several other regions in the genome where we see this pattern, including regions that are known to be important for immune function. However, the bulk of introgressed Neanderthal DNA appears to have been deleterious." As a result, Neanderthal DNA has largely been selected against in the evolution of our species. "Across the traits that we examined there is a consistent pattern of Neanderthal DNA contributing less to these traits than expected, which is the pattern that we would expect if this DNA is deleterious and is being gradually removed from the modern human population," Sankararaman said. The lingering effects of Neanderthal introgression on human complex traits Xinzhu Wei, Christopher R Robles, Ali Pazokitoroudi, Andrea Ganna, Alexander Gusev, Arun Durvasula, Steven Gazal, Po-Ru Loh, David Reich elifesciences.org/articles/80757 |
|

