|
|
Post by Admin on Jul 21, 2021 20:42:01 GMT
High-Density Genetic Variations of Modern and Ancient Genomes Show Fine-Scale Population Structure of Northern Han We additionally investigated the fine-scale genetic structure of Shanxi Han by determining the genetic relationships under the context of 65 worldwide populations (Figure 4A). PCA of worldwide populations allocated Shanxi Han at the end of the Eurasian–American genetic cline (Figure 4B). And Shanxi Han clustered closely with Beijing Han in the finer scale of variations from East Asia (Figure 4C). The observed patterns of genetic affinity were subsequently supported by the results from ADMIXTURE analysis (Figure 5). East Asians were a homogeneous population when the predefined ancestry populations are less than eight. Genetic component kept similar between Shanxi Han and Beijing Han, Denver Chinese, Han, and northern Han. The pairwise Fst genetic distances estimated using SNP data were presented in Table S7. The smallest genetic distance was identified between Shanxi Han and Beijing Han (Fst = 0.0006), followed by Tujia and another northern-Han (Figure 6A). Outgroup-f3 in the form of f3 (X, Shanxi Han; Yoruba) showed that the greater genetic affinity identified between Shanxi Han and Han Chinese populations, subsequently followed by the southern Tai-Kadai and Hmong-Mien speakers, and Western Trans-Himalayan and northern Altaic speakers (Figures 6B, C). We subsequently estimated the D-statistics in the form of D (X, Y; Shanxi Han, Yoruba), where X represents the worldwide populations and Y denotes the Chinese populations from different language families, to explore the status of allele sharing. Our results provided supporting evidence for more shared genetic drift between Shanxi Han and northern-Han or neighboring minorities (Figure 7 and S6 and S7). We used the admixture f3(Source1, Source2; Shanxi Han) to find the potentially admixed ancestral populations. Two hundred sixty-four out of 2,016 pairs were observed with significant negative values (Figure 8). The potential ancestry populations revealed by the admixture-f3 indicated that the ancestral populations of Shanxi-Han derived their ancestry from southern Chinese-related population (Ancestral Southeast Asians) and East-Siberian-related population (Ancestral Northeast Asians), like the two ancestry populations observed in Indian (Ancestral North Indians and Ancestral South Indians) (Reich et al., 2009). FIGURE 4 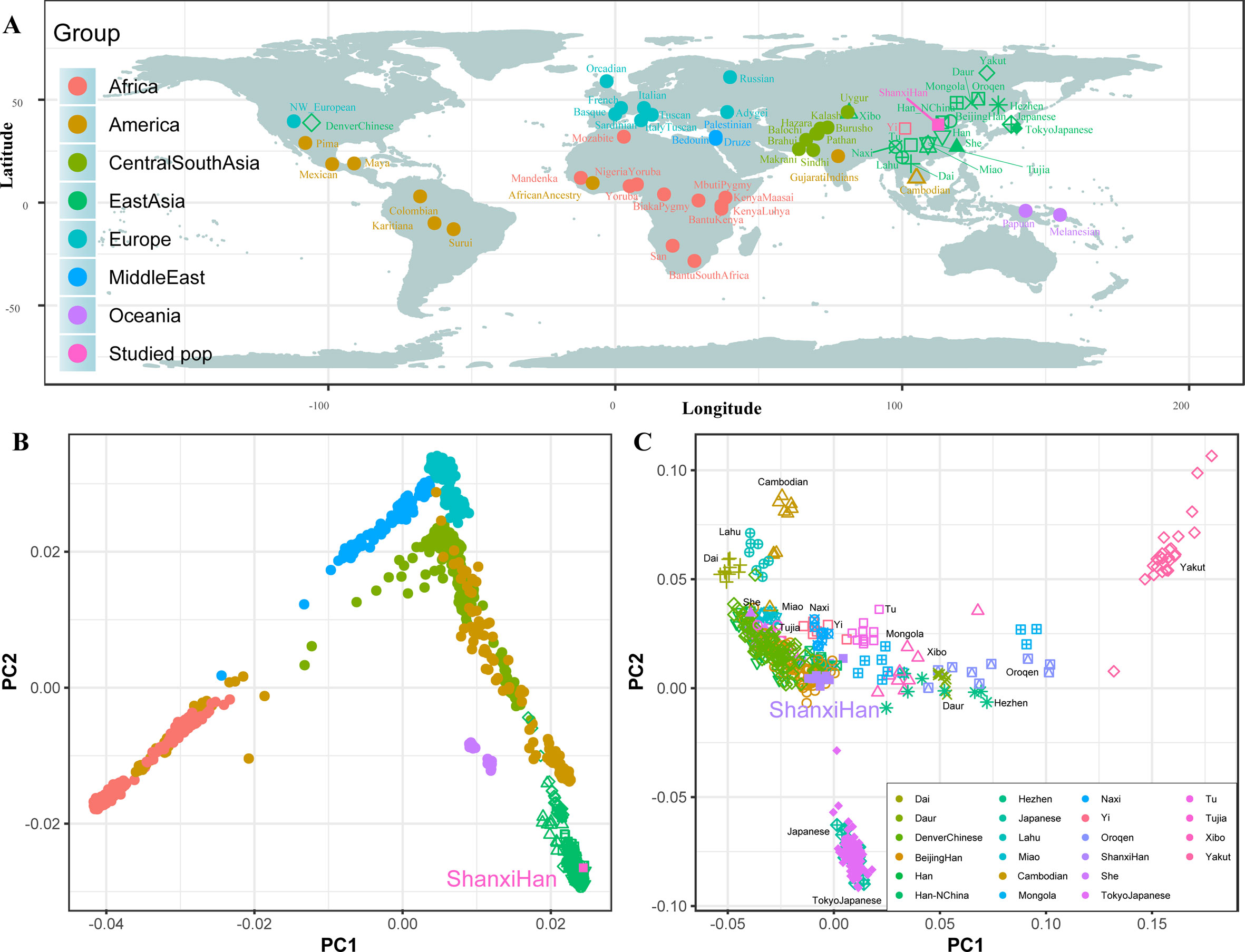 Figure 4 Geographic position (A) and PCA results (B and C) among 65 worldwide populations. 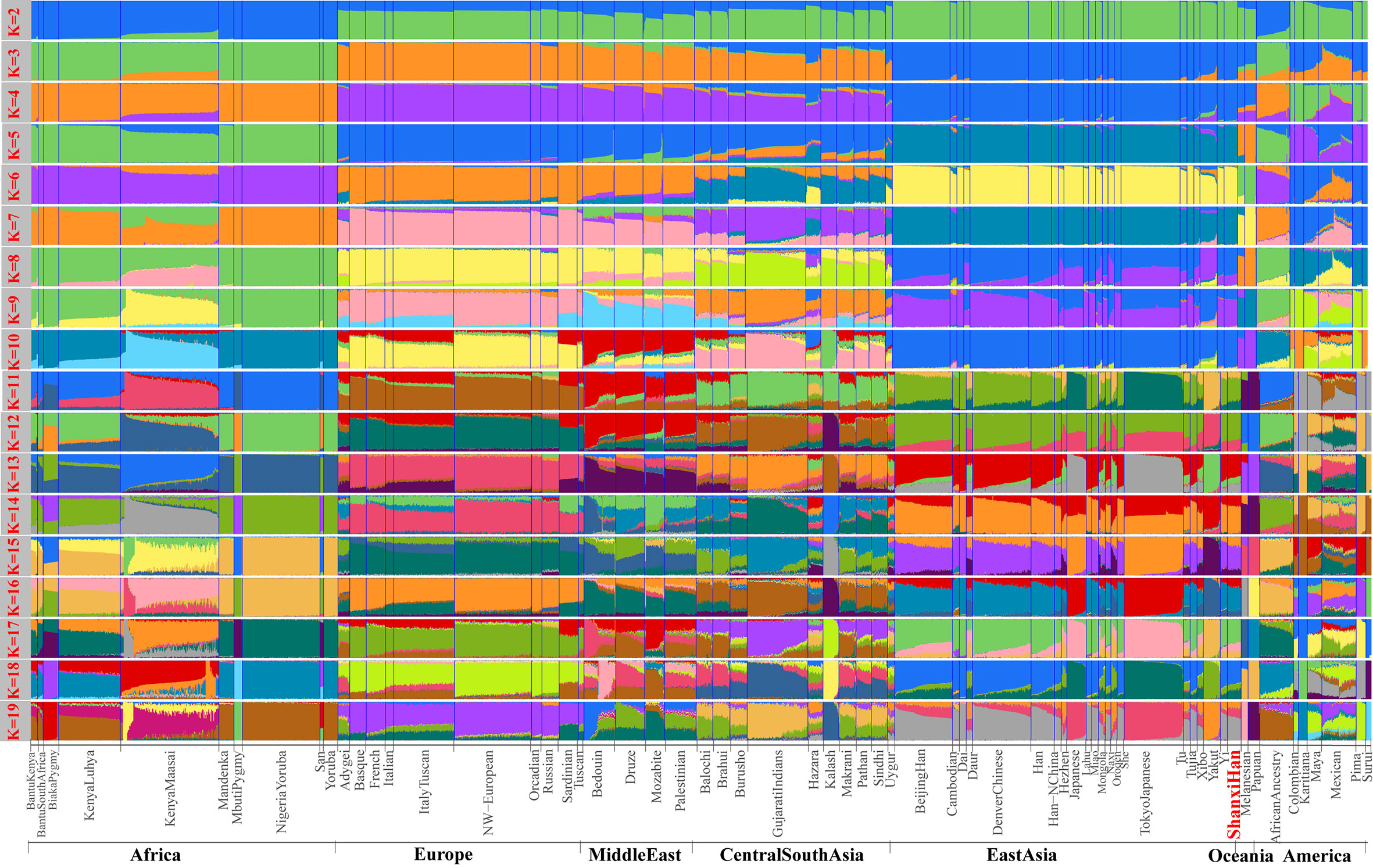 Figure 5 Model-based results of 65 populations with predefined ancestry populations varying from 2 to 19. 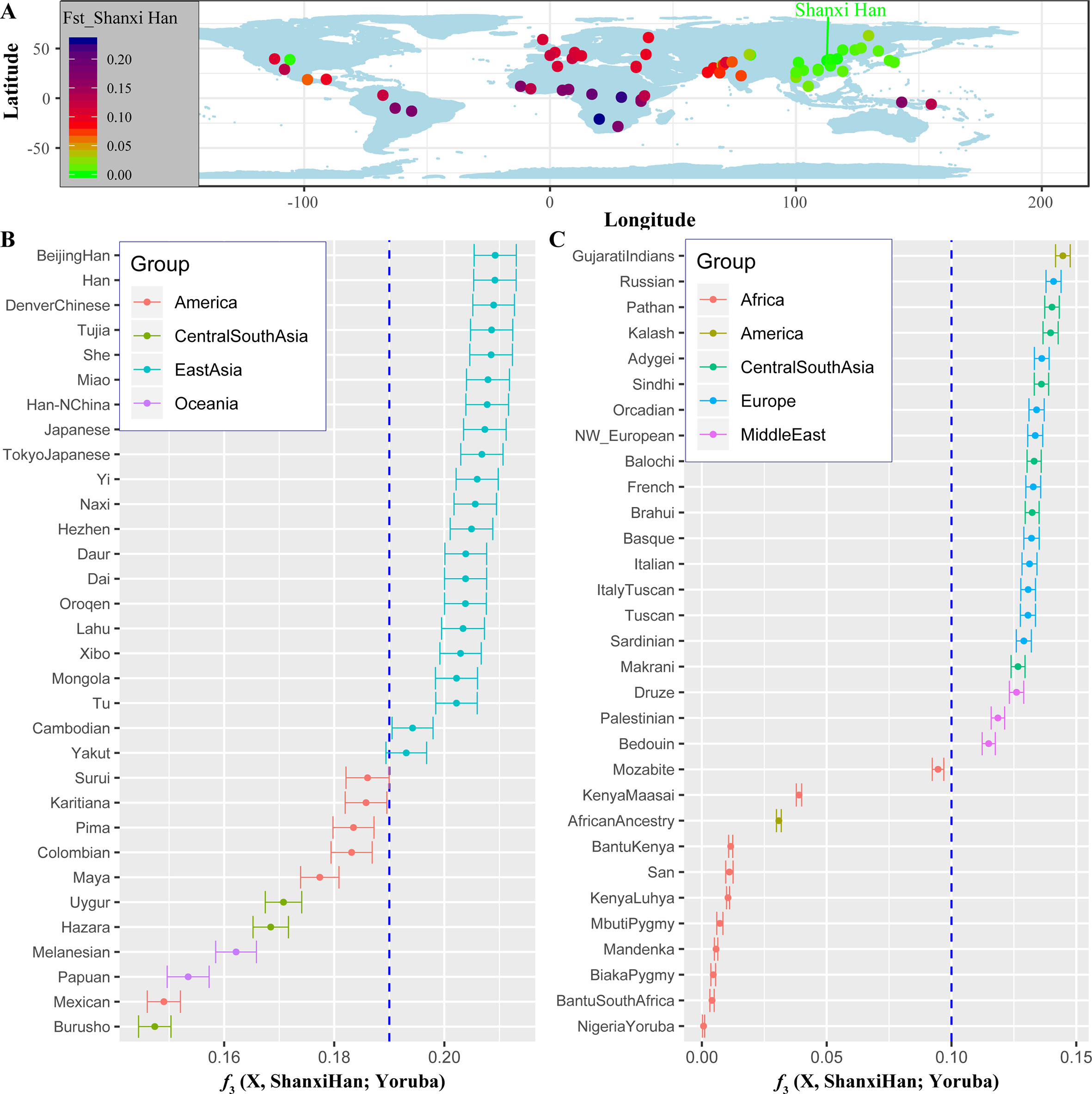 Figure 6 The genetic affinity between Shanxi Han and other 64 worldwide populations revealed by pairwise Fst genetic distance (A), shared alleles (B and C). |
|
|
|
Post by Admin on Jul 21, 2021 21:44:55 GMT
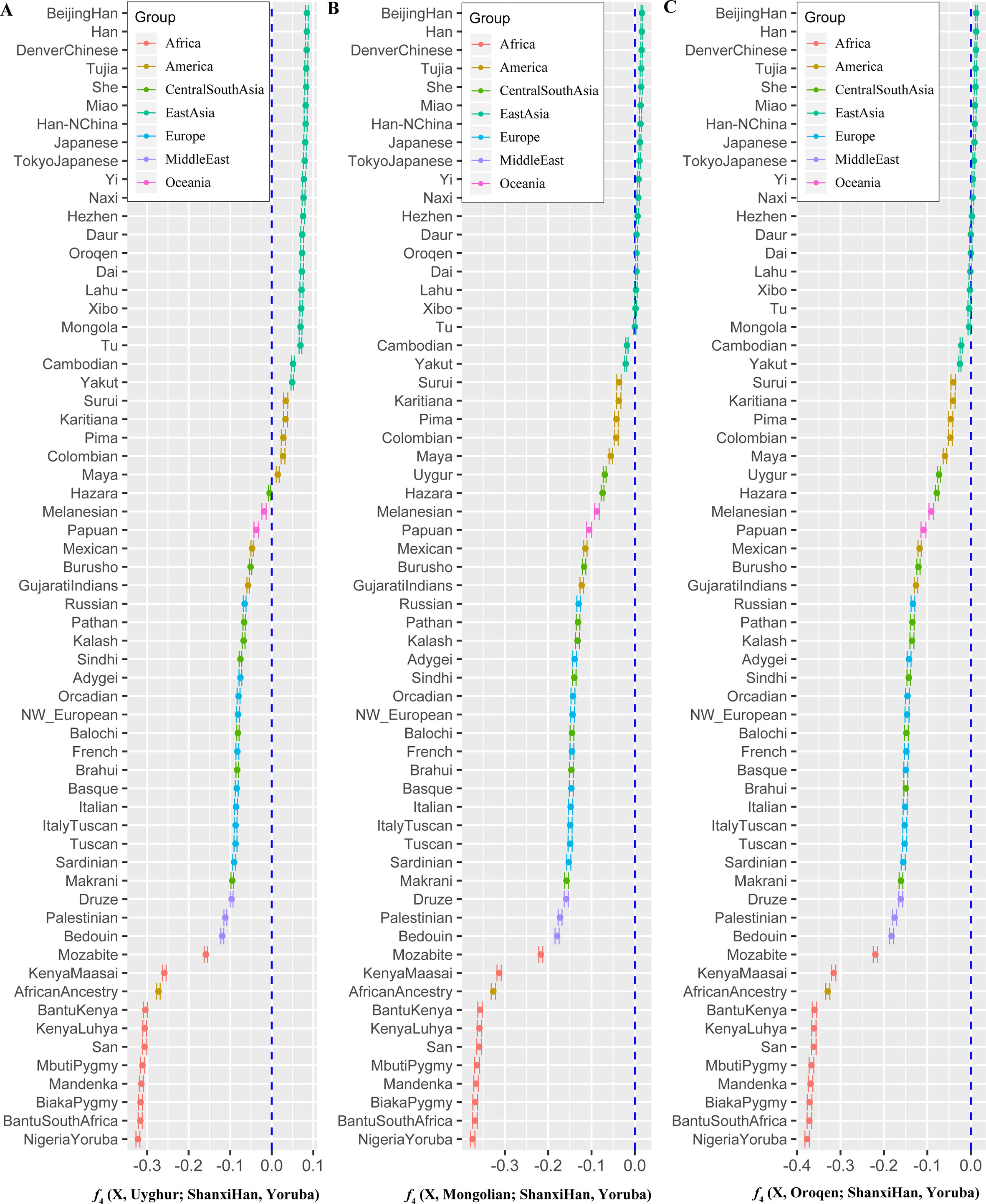 Figure 7 Shared genetic components with Shanxi Han between Altai-speaking populations and other worldwide reference populations: Turkic-speaking Uyghur (A), Mongolic-speaking Mongolian (B) and Tungusic-speaking Oroqen (C). 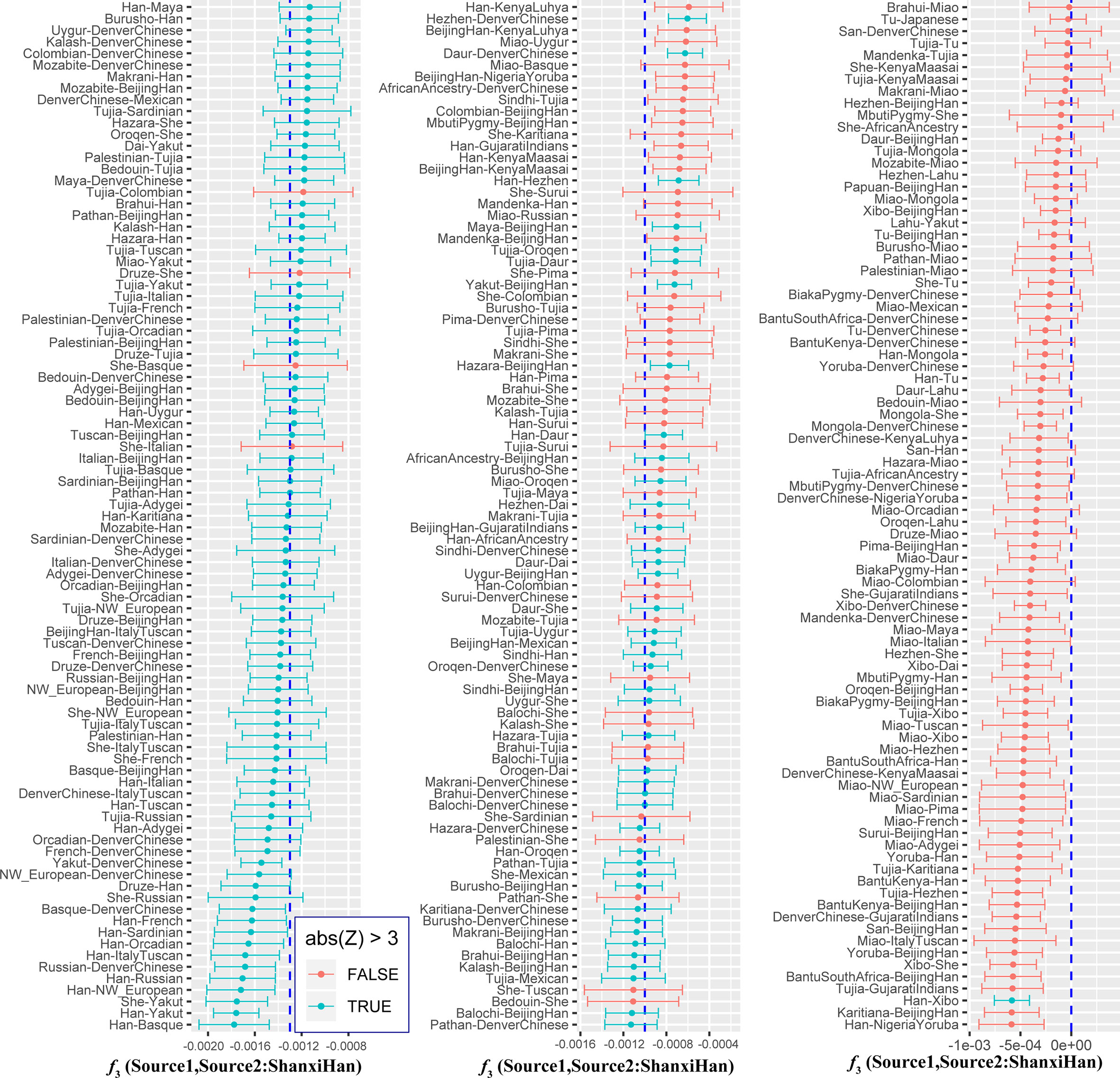 Figure 8 Admixture f3 results with significant negative f3 values. To further validate the minimum streams of ancestry populations and evaluate the corresponding admixture proportion, we first performed the TreeMix analyses among 65 worldwide populations (Figures 9 and S8) and 25 Asian populations (Figure S9). A larger number of recent admixtures or migrations were observed in our TreeMix model. Considering the statistical significance (f3 = −0.0018 and Z = −6.662) was observed in the form of f3(She, Yakut; Shanxi Han), we then conducted the qpWave and qpAdm using the She and Yakut as source populations and using Yoruba, San, Papuan and Melanesian as outgroup populations. QpWave results (p = 0.052 for rank1) indicated that Shanxi Han was derived from two ancestral populations. The qpAdm analysis further suggested that Shanxi Han has derived 25.2% Yakut-related ancestry and 74.8% She-related ancestry. 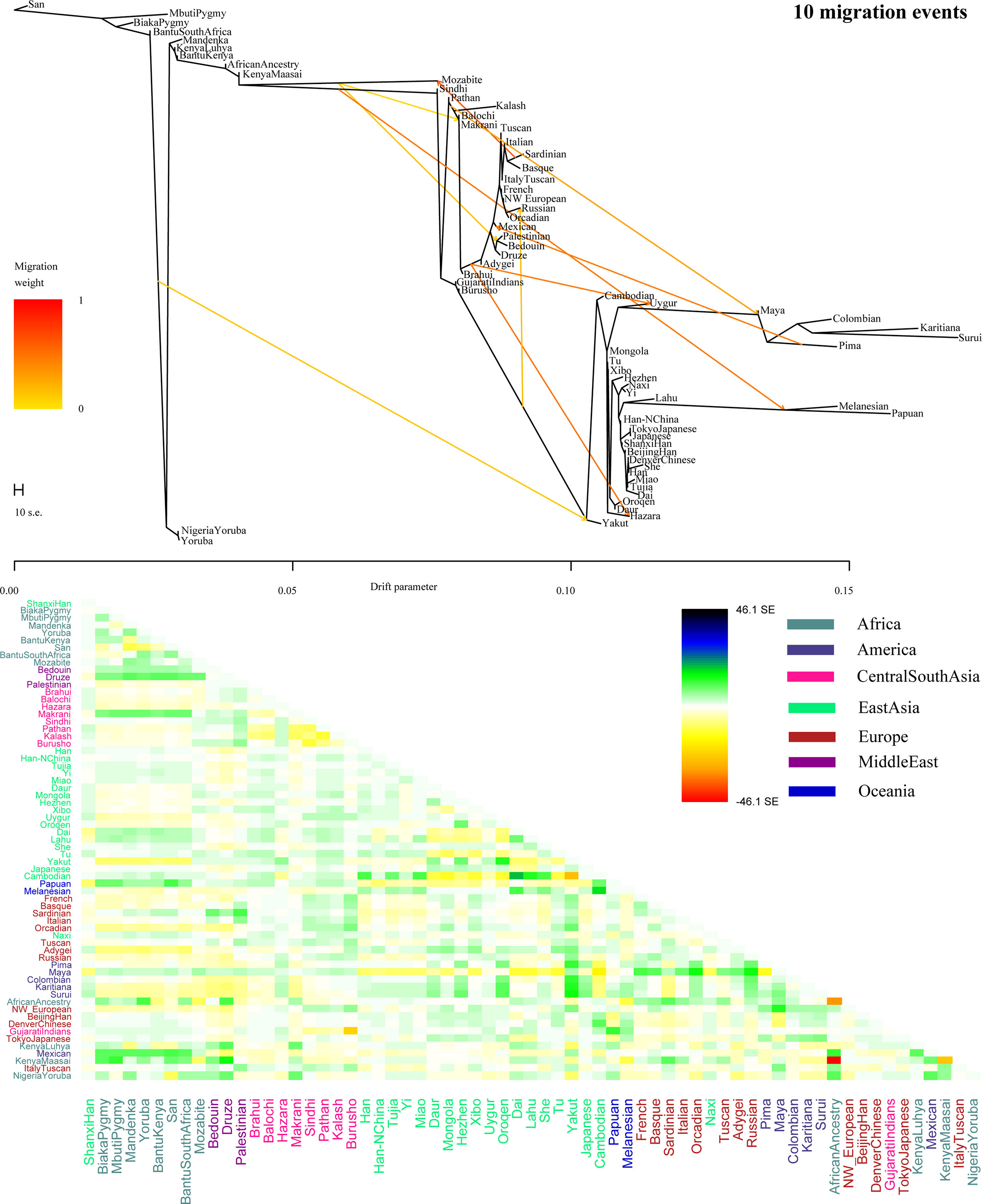 Figure 9 Population splits and admixtures among 65 populations with a prior assumption with 10 migration events inferred from ML tree and model residual. The top panel represents the ML tree with the ten migrations and the bottom panel shows corresponding model residuals. Nineteen ancient populations from Eurasian were employed to explore the genetic admixture history between Shanxi Han. The shared genetic history of all pairs was presented in Figure S10. Denisovan and Vindija Neanderthal shared smaller genetic components with others. We found that Shanxi Han kept a distant genetic relationship with two archaic human populations and Italy Iceman, and shared more alleles with DevilsGate (0.2039) and other ancient populations from Southeast Asia and Nepal (Figure 10). DevilsGate, Oakaie showed significantly negative f3 value in the form of f3(A, B; Shanxi Han) (Table S8). We finally used qpWave to find that the minimum ancient ancestry streams modern northern-Han were 2 (rank1: p = 0.118). QpAdm further showed that DevilsGate Hunter-Gatherer-related population contributed 45% ancestry and Oakaie-related ancient population contributed 55% ancestry to modern northern-Han Chinese. |
|
|
|
Post by Admin on Jul 22, 2021 5:09:52 GMT
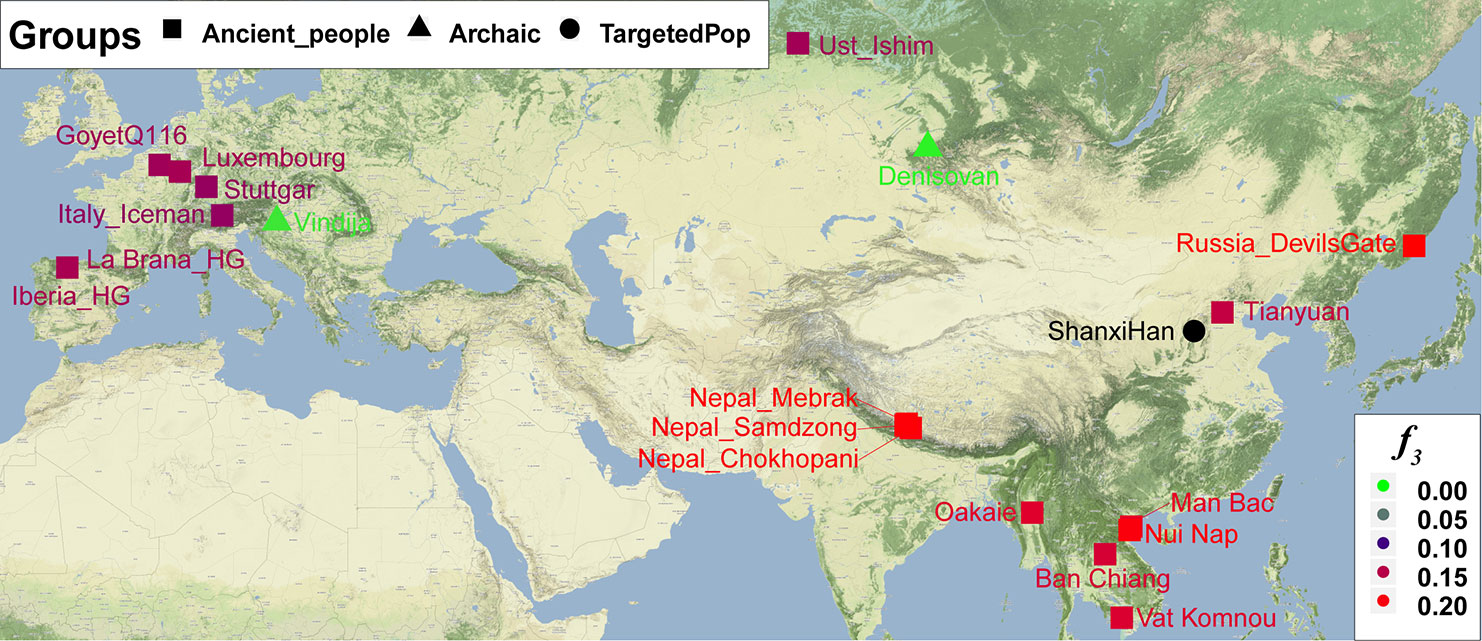 Figure 10 The genetic affinity between Shanxi Han and 19 ancient populations revealed from f3-statistics. Discussion East Asia is occupied by anatomically modern human 50 kya when they migrated out of Africa. These regions are populated by the hunter–gatherer over 40 kya in the Paleolithic time (Nielsen et al., 2017). Under the natural selections from different environments, substance strategies and disease pathogens, the different ethnic group formed their specific genetic structure with a different culture, appearance, and language. In the Neolithic time, agriculture originated from the Yellow, Yangtze and Zhujiang River Basins, may be also included in Liaohe, promoted the process of population genetic structure change with different cultures (Piao et al., 2010). For China in East Asia, the world’s largest ethnic group of the Han Chinese, 55 officially recognized and several unrecognized ethnic groups are subsequently formed with their specific cultural background. The languages they used in this region include over nine language families: Indo-European, Altaic (also called “Trans-Eurasian,” including Turkic, Mongolic, and Tungusic language groups) mainly distrusted in the north; Tai-Kadai, Hmong-Mien, Austronesian, Austroasiatic, and Trans-Himalayan language families in the south. Population substructures among Chinese modern populations revealed by our autosomal STR panel have supported the patterns of population relationship found by the X and Y-chromosome markers, as well as ancestry-informative single nucleotide polymorphisms (He et al., 2017b; He et al., 2018a; He et al., 2018c). Here, our study presented the first comprehensive genetic analysis, including autosomal STR, a meta-analysis of mitochondrial and Y-chromosomal haplogroup distribution, and autosomal SNP data of northern-Han Chinese residing in Shanxi Province. We performed the comprehensive population comparison to investigate the origin, genetic legacy and detailed genetic relationship of modern Han Chinese population, especially for the northern Han Chinese. Our results showed that Altaic and Trans-Himalayan speakers except for Sinitic speakers harbored considerable genetic differences with Han Chinese populations. However, no apparent genetic differentiation between Hmong-Mien-, Tai-Kadai and neighboring Han Chinese populations is revealed in our present study (Figure 1). Analysis from the haplogroup distribution of Neolithic Chinese populations showed that the significant association of genetic continuity between ancient populations from Yellow River Valley sites (Mogou, Taojiazhai, and Hengbei) and modern northern-Han Chinese (Figures 2 and S5). Whole-genome high-density SNP data illustrate that Shanxi Han Chinese inherited 25.2% their ancestry from Yakut-related population and 74.8% from She-related population. Ancient autosomal genetic variation subsequently shows a two-way admixture from ancient North East Asian (45% ancestry from DevilsGate Hunter-Gatherer-related population) and ancient South Asian (55% ancestry Oakaie-related ancient population). These results consistently showed a more complex and ancient population admixture history of northern Han Chinese. Han Chinese may be originated from the admixture between the ancient Tibeto-Burman population and a local pre-Sinitic population which may have been linguistically Altaic in the Neolithic time when agriculture emerged in Yangtze and Yellow River Basins. A recent large-scale whole-genome variation study covering 11,670 Han Chinese individuals from 24 out of 33 administrative regions was carried out to explore the Han Chinese population genetic structure and genetic ancestry (Chiang et al., 2018). Their valuable finding of east-west genetic distinction among Han Chinese is one indispensable previously unrecognized population structure, which is perfectly complemented the north-south differentiation previously found by Xu et al. (2009). Using high-density SNP typing data and other scientists using uniparental markers and classical markers (Xu et al., 2009; Stoneking and Delfin, 2010; Sanchez-Mazas et al., 2011). Our results in this study on the basis of the nationwide STR variations also provide the microsatellite evidence for north-south genetic cline but fail to reveal the east-to-west difference, which may be caused by the sample coverage. Lu et al. (2016) whole-genome sequenced 39 Han Chinese and 38 Tibetan individuals to investigate the gene pool of the Tibetan and Han group. They found that Tibetan and Han Chinese diverged from each other at 7,000–13,000BC during the last glacial maximum (Lu et al., 2016). Wang et al. (2018) recently also tried to investigate and elucidate the precise divergence time, genetic structure and admixture history between Han Chinese and neighboring country populations (Japanese and Korean). Their results suggested that Han Chinese and other focused two populations split approximately 1,000–1,600 BC in the Shang dynasty in Chinese history and subsequently substantial genetic admixture between them and other adjacent populations have occurred (Wang et al., 2018b). The processes of ancient whole-genome DNA studies with the technological innovations of DNA hybridization enrichment and next-generation sequencing has revolutionized the phylogenetic relationship and population history reconstruction in the European, American, Oceanian and even southeast Asians (Nielsen et al., 2017). In East Asia, just two projects respectively focused on one 40,000-year-old individual from Tianyuan cave and two hunter–gatherers from Devil’s Gate have been performed. Yang et al. (2017) sequenced the whole-genome of Tianyuan ancient people (40,000BP) and discovered a strong genetic affinity between these ancient people and present populations, which indicated that there is a genetic continuity or population turnover in the East Asian continent (Yang et al., 2017b). Siska et al. (2017) genome-wide analyzed two Devil’s Neolithic individuals (∼7,700BP) near to the Amur basin and also detected the genetic continuity in northeast Asia. If ancient people from Paleolithic, Neolithic, Bronze, and Iron Ages in East Asia are all sequenced and conducted corresponding population history reconstruction combined with the historical, cultural, linguistic and archeological findings, a complete genetic landscape of the East Asians will be obtained. However, a number of ancient people excavated from different archeological sites in China have so far received little attention. Fortunately, there still some exploratory projects focused on the genetic variations of the uniparental markers (mtDNA and Y-chromosome) and Neolithic or historical ancient people been carried out. Thus, we can perform the first meta-analysis to investigate the phylogenetic relationship between the ancient population and modern northern-Han Chinese population. Our present meta-analysis results from the Neolithic ancient people and modern Han Chinese on the basis of the combined genetic variations of mtDNA and Y-chromosome first showed that the ancient populations from West Liao River Valley sites (Dasanqian and Niuheliang) and Yellow River Valley sites (Hengbei, Taojiazhai) share considerable similar mitochondrial haplogroup with the modern northern-Han Chinese populations. For Y-chromosome variations, ancient people from the Hengbei site shared the more significant genetic similarity with modern northern-Han Chinese from Shanxi and Heilongjiang provinces, and Dashanqian and Mogou ancient people bear a similar genetic assemblage with modern Taiwan Han people. Mogou site in the Ganqing region adjacent to the central plain is the hometown of Di-Qiang people who are thought as the direct ancestral population of Han Chinese, which is genetically close to the Han Chinese population. Our results reveal a close genetic relationship among Hengbei, Mogou and modern northern-Han Chinese populations. Our findings combined with the archeological, historical and linguistic evidence consistently supported the admixed genetic origin of modern Han Chinese. Perspective In summary, we genotyped 23-autosomal-STRs in 3,089 Shanxi northern-Han Chinese individuals and provided the first batch of allele frequency, forensic and population genetic parameters of northern Han Chinese. Comprehensive worldwide and nationwide population comparisons not only showed that Shanxi harbored a strong similar genetic assemblage with adjacent Han populations but also illustrated that there were apparent genetic distinctions between north-to-south Han Chinese as well as genetic differentiation between populations belonging to different language families, obviously differences observed between Tibetan, Uyghur, and others here. The first meta-analysis based on the mitochondrial and Y-chromosomal genetic variations among ancient and modern Asian populations showed a genetic affinity and genetic continuity between Mogou, Hengbei ancient population and present-day northern-Han Chinese. We also found Neolithic agriculture expansion related Dashanqian and Niuheliang ancient populations are genetically close to modern northern Han. The qpWave/qpAdm modeling further revealed that modern northern Han Chinese carried 74.8% She-related ancestry and 25.2% Yakut-related ancestry. Both Hengbei-associated and Tibetan-related uniparental lineage (D haplogroup) were observed in modern Northern Han Chinese. Besides, approximately 45% DevilsGate-like ancestry, one Tungusic-affiliated Neolithic population, was modeled via ancient DNA. Summarily, consistent with previous linguistic and archaeological evidence, the genetic mixing that led to the emergence of a Han Chinese ethnicity occurred at a very early period, probably in Neolithic times, and this mixing involved an ancient Tibeto-Burman population and a local pre-Sinitic population, which may have been linguistically Altaic. Fine-scale population history reconstruction of north Han from modern and ancient genomes consistently model their ancestral populations deriving from ancestral North East Asian and ancestral South East Asian. |
|
|
|
Post by Admin on Aug 3, 2021 6:26:09 GMT
Munda languages are father tongues, but Japanese and Korean are not
Gyaneshwer Chaubey and George van Driem
Over two decades ago, it was observed that the linguistic affinity of the language spoken by a particular population tended to correlate with the predominant paternal, i.e. Y-chromosomal, lineage found in that population. Such correlations were found to be ubiquitous but not universal, and the striking exceptions to such conspicuous patterns of correlation between linguistic and genetic phylogeography elicit particular interest and beg for clarification. Within the Austroasiatic language family, the Munda languages are a clear-cut case of father tongues, whereas Japanese and Korean are manifestly not. In this study, the cases of Munda and Japanese are juxtaposed. A holistic understanding of these contrasting cases of ethnolinguistic prehistory with respect to the father tongue correlation will first necessitate a brief exposition of the phylogeography of the Y chromosomal lineage O. Then triangulation discloses some contours and particulars of both long lost episodes of ethnolinguistic prehistory.
The uneasy relationship between language and the Y chromosome
The observation that the linguistic affinity of the language spoken by a particular population tends often to correlate with the predominant Y-chromosomal lineage found in that population was first pointed out by a Swiss–Italian team of geneticists (Poloni et al. 1997, 2000). As Lendering (2010: 252) observed in his history of Alexander the Great, ‘As so often happened in the wars of antiquity, the widows married the murderers of their spouses’. Historical accounts record that the campaigns led by Genghis Khan, Tamerlane and other conquerors availed themselves of the same tactic, and Kivisild (2014) has wryly qualified warfare as a Y chromosome-linked pathology.
Although the motif of male genocide has repeated itself throughout history, conquering incursive groups might very well have been more clement in many particular instances, yet may nonetheless have benefitted from preferential access to the womenfolk and a more prolific siring of progeny by dint of élite dominance. As we have noted before, the preponderance of such correlations allows us to deduce that a mother teaching her children their father's tongue must have been a prevalent and recurrent pattern in linguistic prehistory. As a consequence of this social and demographic mechanism, paternally inherited polymorphisms often serve as tracers for linguistic dispersals in the past (van Driem 2007), and a particular Asian subset of such patterns of correlation forms the topic of the present paper.
Furthermore, the shallower time depth of the linguistically reconstructible past and the putative age of recognised language families match the time depth attributed to the most recent common ancestor of many geographically widespread paternal lineages (Zerjal et al. 2003; Balaresque et al. 2015). However, a few other forces are hypothesised to influence this topology equally (Karmin et al. 2015). In sharp contrast with our mitochondrial past and also in comparison with the rest of the genome, Y chromosomal phylogeography is relatively recent, having undergone a global bottleneck towards the end of the last ice age, when a subset of the then existing paternal clades began eradicating and out-competing other clades (Karmin et al. 2015; Silva et al. 2017). Effective palaeolithic founder populations of major modern paternal subclades were small. In terms of Y-chromosomal lineages, entire new populations arose from small surviving subsets that had passed through such bottlenecks. Similarly, many of today's language families and linguistic phyla appear to be the result of prehistoric bottlenecks.
When languages and genes happen to exhibit a correlation, such a relationship ought not to be confused with identity, and a chromosomal marker should not be simplistically equated with populations speaking languages of a particular linguistic phylum. Rather, markers on the Y chromosome serve as proxies or tracers for the movements of paternal ancestors. Although ubiquitous, the father tongue correlation is not universal. Exceptions to the father tongue correlation, such as Hungary and Baltistan (van Driem 2007), are not unique cases. Rather, the meticulous study of correlations of genetic polymorphisms with the geographical distribution of language families and their constituent subclades as well as the discrepancies between genetic and linguistic phylogeography, nowadays enriched with the information rendered available through whole genome studies and ancient DNA findings, are providing us with an increasingly differentiated view of the past with ever greater detail.
|
|
|
|
Post by Admin on Aug 3, 2021 22:13:20 GMT
The East Asian linguistic phylum The East Asian linguistic phylum was proposed by Starosta in Périgueux in 2001, one year before his death. His thinking was published posthumously, shown in Figure 1 (Starosta 2005; van Driem 2005). In proposing to unite the Kradai, Austronesian, Trans-Himalayan, Hmong-Mien and Austroasiatic language families into a single East Asian linguistic phylum, Starosta had numerous precursors. Conrady (1916, 1922) and Wulff (1934, 1942) proposed a linguistic phylum consisting of Austroasiatic, Austronesian, Kradai and Trans-Himalayan, whilst Benedict (1942), Blust (1996) and Peiros (1998) proposed an Austric superfamily comprising Austroasiatic, Austronesian, Kradai and possibly Hmong-Mien. The more modest proposal to unite just two of these five East Asian language families, viz. Kradai and Austronesian, was first advanced by Schlegel (1901, 1902) and then seconded by Benedict (1942). However, the first sound historical comparative evidence for the Austro-Tai family was adduced by Ostapirat (2005, 2013). 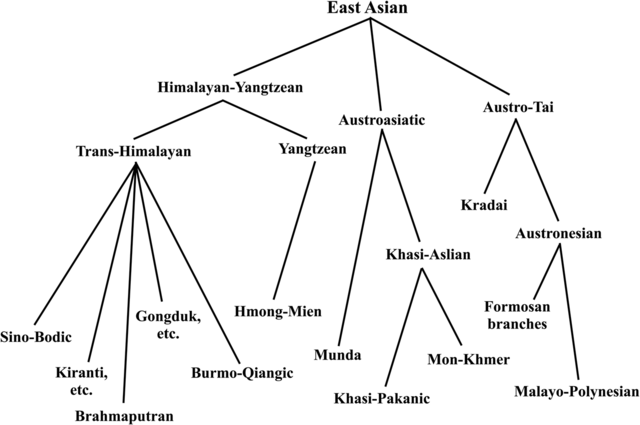 Figure 1. The enhanced 2012 Benares recension of Starosta's East Asian linguistic phylum (Starosta 2005; van Driem 2014). For his grander East Asian linguistic phylum, Starosta adduced the putative shared morphological vestiges of an agentive prefix *<m->, patient suffix *<-n>, instrumental prefix <s-> and perfective prefix *<n->. A discussion of the merits of this evidence strikes us as being of little utility, since we consider the antiquity of the proposed linguistic phylum to lie at the ‘linguistic event horizon’ or maximal time depth reconstructible through methodologically sound historical linguistic comparison. Beyond this epistemological boundary hypotheses regarding long-distance linguistic relationship are reduced to sheer speculation. Rather, Starosta (2005: 194) modestly proposed that the ‘potential utility’ of his hypothesis lay ‘in helping to focus scholars’ efforts on particular specific questions, resulting in the replacement of parts of this hypothesis with better supported arguments’. Following Ostapirat, Starosta's East Asian linguistic phylum may be construed to comprise four recognised language families: Austro-Tai, Trans-Himalayan, Hmong-Mien and Austroasiatic. |
|

