|
|
Post by Admin on Jan 17, 2019 21:27:48 GMT
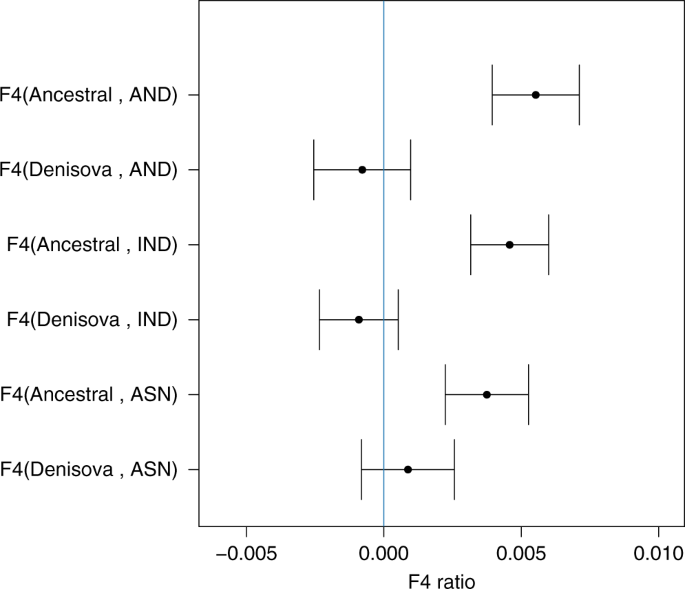 Fig. 1 Increase of Neanderthal ancestry in Asians compared to Europeans. The introgression amount is calculated by the F4 ratio test. F4 (Outgroup, Altai; Europeans, X)/F4(Outgroup, Altai; Africa, Vindjia) where Outgroup can be either Denisova or Ancestral alleles from 1000 Genome and X is either East Asian (ASN), Indian Tribal (IND) or Andamanese (AND). In the y axis to make it short we only mention F4(Outgroup, X) as the other populations remain constant for all the F4 ratio tests. In the x axis we have the amount of F4 ratio and the standard error is denoted by bars (standard error was calculated by Jackknife method using 555 blocks). The blue line signifies 0 value for F4 ratio test ABC-DL analysis We considered several plausible competing demographic models which describe scenarios that have been proposed by different studies to explain the observed genetic diversity of introgression patterns among current old world AMH populations, Neanderthals and Denisovans (see Fig. 2 and Supplementary Table 5). The implemented demographic models are variations from an initial model with many accepted features (Model A): OOA origin of modern humans, with a Eurasian split between Europeans and the group comprising two subgroups, East Asians, Indian and Andamanese on one hand, and Papuans and Australians on the other. Introgression of Neanderthals is found in all OOA populations, and that of Denisovans in the Oceanian populations. Introgression from an extinct group of distant hominins into Denisovans14,16 has been considered in all models, and this introgression does not affect directly our core results. From this basic model, several variations have been proposed to better fit the genetic data; here we consider the following. Model B: decrease of European Neanderthal ancestry due to admixture with a modern human, “basal Eurasian”, a ghost population non-introgressed by Neanderthals (Xn)34. Model C: two introgression events from Neanderthals, the first affecting all OOA populations and the second only affecting Asia35,36. Model D: two introgression events both from Neanderthals and Denisovans, with a Denisovan introgression on Asians before the split of Oceanian populations14,16. The remaining models have a single introgression of Neanderthals and Denisovans (as in Model A) but with an introgression to Asian and Oceanian populations from an extinct (Xe) hominin population that is a sister group of Neanderthals (Model E), or is a sister group of Denisova (Model F;20), or is an outgroup of both Neanderthals and Denisovans (Model G;6). Given the recently discovered Neanderthal–Denisovan hybrid37, we also considered a scenario in which this extinct hominin is an admixed population between Neanderthals and Denisovans (Model H)37, a similar model of a trichotomy as will be seen below. Furthermore, Model E and Model F can be considered particular cases of Model H when the percentage of Denisovan admixture is 0 or 100%, respectively. In all the models, the basic tree structure remained constant although all the demographic parameters—including the amount of introgression- were estimated. 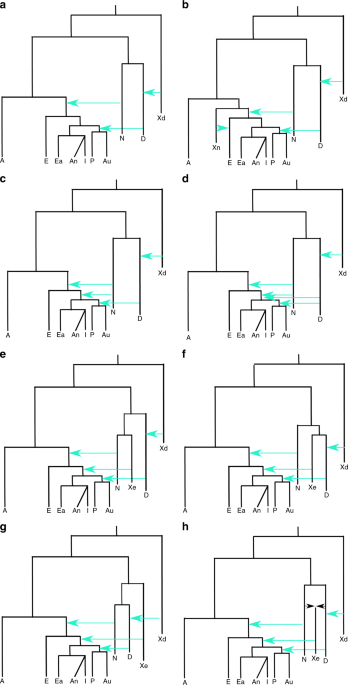 Fig. 2 Demographic models implemented for explaining the genetic variation present in current African (A), European (E), East Asian (Ea), Andamanese (An), Indian (I), Papuan (P) and Australian (Au) populations, in relation with Altai Neanderthal (N) and Denisovan (D) archaic populations. The process of archaic introgression in Asian populations is modelled either by different introgression events from a anatomically modern human ghost population (Xn), b, c, d known archaic populations such as N and D and or e, f, g, h by an unknown archaic extinct (Xe) population. An archaic population (Xd) introgressing to Denisovans is also modelled. Turquoise arrows indicate single pulse events of archaic introgression. All models include recent continuous migrations between African and European, European and East Asian, and Papuan and Australian populations We first evaluated the performance of the proposed ABC-DL methodology for distinguishing between the proposed eight models. We used simulated data as pseudo-observed data and run the full ABC-DL pipeline; we applied a ‘hard’ model classification to assign the simulated data to the model with the highest posterior probability (see Supplementary Note 2). The confusion matrix (Supplementary Table 6) summarizing the model classification shows that the proposed ABC approach identifies the model that generated the data (Modelsim) with posterior probability P(Modelsim = Modelabc | Data) > 50% in all the models. When comparing the eight considered models by ABC-DL with the observed data, the ABC model inference supports model H (P(Model = H|Data) = 0.46), namely the introgression of genes from an archaic Denisovan-Neanderthal admixed extinct population (Xe) into Asian AMH populations, before the split of Oceanian groups, closely followed by Model F (P(Model = F|Data) = 0.38), which models Xe as a sister population of Denisovans. Model H is slightly more likely than Model F (1.2 times more likely), and much more likely than models E (5 times more likely), G (7.6 times) and D (107 times). Models not considering the presence of an extinct archaic ‘ghost’ population introgressing AMH -A, B, C and D- obtained a posterior probability of ~0 (see Supplementary Figure 4). Therefore, only models with admixture of an extinct population in the Neanderthal–Denisovan clade, but with significant differences to each of them, are supported by the ABC-DL framework.  Next, we implemented ABC-DL to estimate the posterior distribution of each parameter for Model H (Fig. 3b and Supplementary Figure 5). Given that Model F is also similarly supported, for completion we also estimated the posterior distribution of the parameters in this model (Fig. 3a). As internal check of the performance of the DL in Model H, we first estimated the Pearson correlation between the parameter values used for the simulation and the predicted parameter value by the DL (Supplementary Table 7). For almost all the parameters from the Model H, we obtained a high correlation between the real parameter and the predicted one by the DL with the exception of the effective population sizes of the unknown ghost populations, which is a very acceptable expectation as we do not have direct information of the genetic variation in these populations. When applied to the observed data (Table 2 and Supplementary Table 8), our ABC-DL approach for estimating parameters of Model H suggests that (i) the introgression of Neanderthals with AMH out of Africa was 1.3% (CI from 0.18% to 2.5%) and occurred 69 Kya (CI from 56 Kya to 88 Kya; assuming a generation time of 29 years38), whereas the split between African and OOA populations took place 121 Kya (CI from 78 Kya to 167 Kya); (ii) the Denisovan introgression to Oceanian populations was 1.6% (CI from 0.4% to 2.5%) and took place 43 Kya (CI from 29 Kya to 50 Kya); finally, (iii) the estimated amount of introgression in the AMH ancestor of current Asian populations by the extinct archaic population Xe was 2.6% (CI from 0.7% to 4.6%) at 51 Kya (CI from 45 Kya to 58 Kya). According to our analyses, Xe appeared 304 Kya (CI from 211 Kya to 375 Kya) from an admixture event of Denisovans and Neanderthals that would have occurred only 14 Kya after the divergence of Denisovans and Neanderthals (314 Kya; CI from 300 Kya to 343 Kya). The estimated divergence between Denisovan and Neanderthals is relatively recent compared to the estimated in16 (381–473 Kya when considering a mutation rate of 0.5e-09 per bp per year or 1.45e-08 per generation assuming 29 years by generation) but higher than in16 (190–236 Kya when considering a mutation rate of 1e-09 per bp per year or 2.9e-08 per generation). Since the number of non-shared variants depends on the time of divergence, the genetic drift at each population and the mutation rate, a possible explanation for the differences in time of divergence could be the mutation rate, which substantially variate among studies. Demographic parameters estimated for Model F are similar in magnitude to the ones estimated in Model H, with credible intervals that overlap the ones obtained for Model H (Supplementary Table 9). 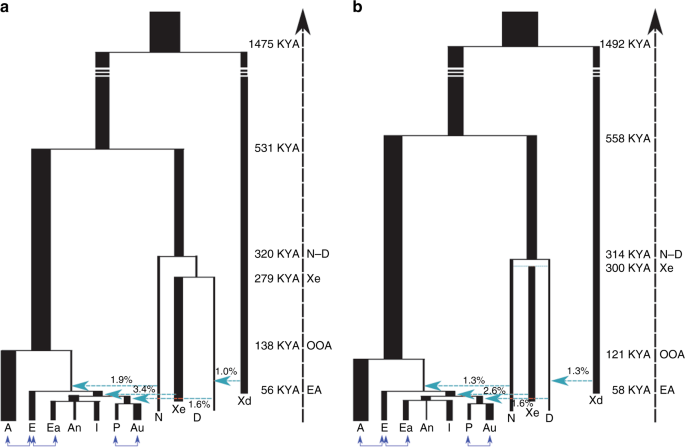 Fig. 3 In order to see whether the proposed Model H fit observed D-statistics, we generated simulations using the mean of the estimated posterior probability of each parameter, and computed D-statistics assuming different population tree topologies for stressing the impact of archaic introgression in Asian populations. Simulation results from ABC-DL for Model H replicate the empirical results of the D-statistics (Table 1, last column) by reproducing the excess of Neanderthal and Denisova ancestry in Asian populations compared to Europeans. Taken together, we have an overwhelming support for the existence of a third extinct branch of the Neanderthal–Denisovan clade; considering the credible interval of the time of split between the archaic populations, a model with a trichotomy is a good consensus for Model D-H as in all the models the Xe separates from Neanderthal or Denisova at a similar time depth of Neanderthal and Denisova split (Supplementary Table 10). The ABC-DL analysis supports that all Asian and Oceanian populations, including East Asians, received an introgression from an unknown extinct archaic hominin population. However, the exact relationship of this extinct hominin population with the other known archaic populations is not completely disentangled. The two models mostly supported by our ABC-DL model comparison suggest that Xe corresponds to one of the two sister-Denisovan lineages (Model F) or this hominin was closely related to the Neanderthals and Denisovans with a slightly greater similarity to Denisovans than to Neanderthals (Model H). In our analyses, Model H is slightly better supported than Model F. Nevertheless, the Bayes factor between Model H and F is so close to one that the difference in the posterior probability of the two models is hardly worth mentioning39. Furthermore, in both models ABC-DL parameter estimation suggests that Xe had a similarly long independent evolutionary history (Table 2, Supplementary Table 8, 9 and 10). Nature Communications volume 10, Article number: 246 (2019) |
|
|
|
Post by Admin on Apr 2, 2019 17:33:13 GMT
The Denisovans were a species or subspecies of humans that are believed to have lived in Asia and Southeast Asia. Little physical evidence of their existence has ever been found, all of it in Denisova Cave in the Altai Mountains in Siberia. But much more evidence of them exists in our genes. Like Neanderthals, the Denisovans interbred with humans. It is currently believed that as humans migrated out of Africa, they encountered and mated with Neanderthal—as they moved farther east, they encountered and mated with the Denisovans. In this new effort, the researchers report evidence suggesting Denisovans mated with humans possibly as recently as 15,000 years ago, in Papua New Guinea. The researchers collected tissue samples from 161 people (representing 14 groups) in Indonesia and Papua New Guinea and sequenced their DNA. They report that in the samples from people in New Guinea, they found evidence of Denisovan DNA from populations that were different from those living in the cave in Siberia. They describe them as D1 and D2 (the population in Siberia is described as D0). They report further that D1 and D2 were so distantly related to D0 that they had to have diverged at least 283,000 years ago. They also found that D2 was so distant that it likely split off approximately 363,000 years ago. The researchers suggest such distant divergence makes D1 and D2 as different from D0 as they were from Neanderthals. They also suggest that D2 might even have to be reclassified to give the group its own name. And finally, they reported that they had found evidence of Denisovans interbreeding with modern humans between 30,000 and 15,000 years ago. Geneticists have been able to identify their DNA and this is helping us to better understand this extinct species of humans. Researchers had been able to establish their genetic legacy in a population ranging from “the Philippines and New Guinea to China and Tibet have inherited 3% to 5% of their DNA from Denisovans” reports the Science Mag . It seems that this branch of the human family left Africa sometime before Homo sapiens and that they lived in Asia for thousands of years. When modern humans entered Asia some 50,000 years ago, members of the two species mated with each other. The Science Mag reports that as a result of this extensive interbreeding many modern Melanesian populations in the Pacific have up to “3% to 5% of their DNA from them.”  A team of biologists led by Murray Cox and Herawati Sudoyo found traces of two types of DNA from Denisovan populations in samples of Papuans from New Guinea. After studying the results using statistical analysis they found that some of the genes had only entered the human genome as late as 15,000 years ago. Finding Denisovan genetic material at so late a date was very exciting. The genetic analysis of the sample from New Guinea adds to the body of evidence for mating between modern humans and the Denisovans. This possibly indicates that isolated populations of Denisovans survived as late as 15,000 years ago in the Pacific Islands . The Science Mag quotes Christ Stringer of the London Natural History Museum as stating that a “late surviving lineage [of Denisovans] could have interbred with Homo sapiens .” Moreover, it suggests that modern humans and the extinct species were mating over a period of tens of thousands of years.  There are those who reject this and, while accepting that while Homo sapiens and Homo denisova may have mated regularly, claim it is highly unlikely that they did so as recently as 15,000 years ago. The discovery may be accounted for by two groups who had been isolated from each other and who both had some Denisovan DNA. Because of migrations they encountered each other after a long period of time and mated and this could explain the findings of the international team of biologists. The evidence from New Guinea is adding to our understanding of the long-extinct species. They were a diverse population that appears to have interbred with modern humans for an extended period. This is changing how we see modern people and how they came to dominate the world, they may have peacefully absorbed groups like the Denisovans by interbreeding. This could also explain why Homo sapiens are the only surviving human species. |
|
|
|
Post by Admin on Apr 12, 2019 18:05:38 GMT
The Indonesian Genome Diversity Project (IGDP) has been run by the Eijkman Institute of Molecular Biology in Jakarta for over a decade, with the goal of capturing a representative sample of genomic diversity across this understudied region. Spanning a transect of communities across the Indonesian archipelago and neighboring regions of ISEA, populations chosen for whole-genome sequencing were selected to reflect the main axes of genomic variation observed in an earlier population genetic study (Hudjashov et al., 2017). We sequenced complete genomes to >30× coverage for 161 individuals, from Sumatra in the west to New Britain in the east (Figure 1). We combined this new dataset with 317 additional high-coverage human genomes sampled world-wide, including those few genomes previously available for ISEA and Oceania (Malaspinas et al., 2016, Mallick et al., 2016), and three complete archaic hominin sequences, the Altai and Vindija Neanderthals (Prüfer et al., 2014, Prüfer et al., 2017) and the Altai Denisovan (Meyer et al., 2012) (Figure S1; Table S1; STAR Methods S1–S5).  Figure 1 Modern and Archaic Ancestry (A) Sampling locations of groups. Outer rims indicate modern ancestry components: blue, Papuan; dark gray, Asian (Table S2; STAR Methods S6). Inner pie charts indicate archaic introgression estimates: green, unambiguous Denisovan introgression; yellow, unambiguous Neanderthal introgression; light gray, introgression consistent with either Neanderthal or Denisovan ancestry (Table S3; STAR Methods S9); the total area of each pie chart corresponds to the total amount of unambiguous Denisovan introgression signal, with Papua 13.7-fold greater than West Eurasia. (B) Principal-component analysis (PCA) of the new dataset including non-African reference samples, showing two major axes of variation—a “Papuan” axis stretching from New Guinea Papuans-New Britain Baining to West Eurasians, with ISEA intermediates, and an “Asian” axis stretching from Papuans-West Eurasians to East Asian samples (STAR Methods S2).  Figure S1 Flowcharts of Analyses, Related to STAR Methods To confirm that the dataset captures expected genomic patterns, we calculated principal components and determined local ancestry along the genome (Dias-Alves et al., 2018) (Figure 1; Table S2; STAR Methods S2 and S6). We observed key features of population diversity in the region, notably a strong cline in Asian to Papuan ancestry across the archipelago with an abrupt transition within the island zone of Wallacea (Cox et al., 2010, Hudjashov et al., 2017). These signals primarily reflect recent events of regional history, particularly the agricultural expansion of Austronesian-speaking populations from ca. 4500 ya. This cline serves, however, as an important backdrop to facilitate understanding of regional signals of genetic contact between anatomically modern humans and archaic hominins. Combining Methods Identifies High-Confidence Denisovan Introgression Because individuals in ISEA carry ancestry from both Neanderthals and Denisovans, these archaic signals must be disentangled. Assigning clear ancestry is a major challenge, especially for single variants or small ancestry blocks with few informative variants, because of extensive shared polymorphisms between the two archaic groups as well as incomplete lineage sorting due to the shared early history of Neanderthals and Denisovans. One way to overcome this problem is with haplotype methods to detect longer introgressing blocks, which have more easily assigned ancestry and are less likely to result from incomplete lineage sorting. Examining introgressing haplotypes of Denisovan ancestry offers further advantages over site-by-site methods such as D statistics (Patterson et al., 2012), because haplotypes provide additional information on introgression dates and better resolution of detailed relationships between introgressing and archaic genomes. To obtain a set of high confidence blocks, rather than all possible stretches of Denisovan introgression, we developed a protocol to extract archaic regions using the intersection of three different statistical methods (Table S3; STAR Methods S7 and S8). Denisovan blocks were classified based on the overlap of (1) ChromoPainter (CP) (Lawson et al., 2012), which was used to identify haplotypes that are more similar to the Denisovan genome than to a panel of sub-Saharan Africans; (2) an updated Hidden Markov Model (HMM) (Racimo et al., 2017, Seguin-Orlando et al., 2014) detecting the same signature; and (3) S∗ (Vernot et al., 2016), which identifies clusters of linked non-African variation. These haplotypes were then filtered, using a range of protocols (Figures 2A–2C; STAR Methods S9a), to remove blocks that were also similar to the Altai Neanderthal (Prüfer et al., 2014) and optionally the Vindija Neanderthal (Prüfer et al., 2017), as measured by CP, leaving a dataset of high-confidence introgressed Denisovan regions.  Figure 2 Effect of Block Filtering on the Denisovan Signal and the Correlation of the Denisovan Signal with Papuan Ancestry To confirm that the dataset captures expected genomic patterns, we calculated principal components and determined local ancestry along the genome (Dias-Alves et al., 2018) (Figure 1; Table S2; STAR Methods S2 and S6). We observed key features of population diversity in the region, notably a strong cline in Asian to Papuan ancestry across the archipelago with an abrupt transition within the island zone of Wallacea (Cox et al., 2010, Hudjashov et al., 2017). These signals primarily reflect recent events of regional history, particularly the agricultural expansion of Austronesian-speaking populations from ca. 4500 ya. This cline serves, however, as an important backdrop to facilitate understanding of regional signals of genetic contact between anatomically modern humans and archaic hominins. Combining Methods Identifies High-Confidence Denisovan Introgression Because individuals in ISEA carry ancestry from both Neanderthals and Denisovans, these archaic signals must be disentangled. Assigning clear ancestry is a major challenge, especially for single variants or small ancestry blocks with few informative variants, because of extensive shared polymorphisms between the two archaic groups as well as incomplete lineage sorting due to the shared early history of Neanderthals and Denisovans. One way to overcome this problem is with haplotype methods to detect longer introgressing blocks, which have more easily assigned ancestry and are less likely to result from incomplete lineage sorting. Examining introgressing haplotypes of Denisovan ancestry offers further advantages over site-by-site methods such as D statistics (Patterson et al., 2012), because haplotypes provide additional information on introgression dates and better resolution of detailed relationships between introgressing and archaic genomes. To obtain a set of high confidence blocks, rather than all possible stretches of Denisovan introgression, we developed a protocol to extract archaic regions using the intersection of three different statistical methods (Table S3; STAR Methods S7 and S8). Denisovan blocks were classified based on the overlap of (1) ChromoPainter (CP) (Lawson et al., 2012), which was used to identify haplotypes that are more similar to the Denisovan genome than to a panel of sub-Saharan Africans; (2) an updated Hidden Markov Model (HMM) (Racimo et al., 2017, Seguin-Orlando et al., 2014) detecting the same signature; and (3) S∗ (Vernot et al., 2016), which identifies clusters of linked non-African variation. These haplotypes were then filtered, using a range of protocols (Figures 2A–2C; STAR Methods S9a), to remove blocks that were also similar to the Altai Neanderthal (Prüfer et al., 2014) and optionally the Vindija Neanderthal (Prüfer et al., 2017), as measured by CP, leaving a dataset of high-confidence introgressed Denisovan regions. As a further check, we compared the total amount of our high-confidence haplotypes with Denisovan ancestry proportions calculated with counting statistics as reported for the Simons Genome Diversity Project (SGDP) samples (Mallick et al., 2016) (STAR Methods S9b). The total introgression in West Eurasia estimated by CP alone did not correlate with genome-wide estimates of Denisovan introgression estimated by D statistics but instead correlated strongly with estimates of Neanderthal introgression (extracting 20% of the signal). Strikingly, however, this signal drops to 2% for our high-confidence Denisovan blocks, showing that Neanderthal spillover has been almost entirely removed from the high-confidence Denisovan block set by our multi-step filtering approach. A strong correlation between Denisovan and Papuan ancestry (r2 = 0.98, p = 2.6 × 10−20) confirms that these two components have interconnected histories (Figure 2D; STAR Methods S9d). In this correlation, a gradient close to 1 and a correspondingly low intercept of –0.01 is observed for the high-confidence Denisovan blocks, consistent with the inference that Denisovan introgression is largely confined to Papuans. Low levels of Papuan ancestry in West ISEA (<5%) match the limited Denisovan introgression observed in the region (Figure 1A). |
|
|
|
Post by Admin on Apr 12, 2019 19:33:15 GMT
Denisovan Populations Introgressed into Papuans Twice To determine whether there is structure within the Denisovan sequences, we calculated mismatch distributions between our high-confidence Denisovan blocks and the high-coverage Altai Denisovan genome (STAR Methods S10). Small ancestry blocks can be a problem for mismatch analysis, because the mismatches of individual haplotypes have an imprecise correspondence to genetic divergence caused by the low resolution offered by a small number of stochastic, discrete polymorphisms (STAR Methods S10a) and because small blocks are more often affected by incomplete lineage sorting. We therefore profiled mismatches across a range of block lengths in Papuans, the population in our dataset with the highest Denisovan introgression. Intriguingly, we observe two clearly separate mismatch peaks in Papuans (Figure 3A; STAR Methods S10a). This suggests that Papuans carry lineages from two genetically different Denisovan populations that had been separated from each other for a very long time. These peaks are also observed when using just the CP blocks or just the HMM blocks alone (STAR Methods 10b). Resolution of the two peaks (here called D1 and D2) improves with block sizes greater than approximately 130 kb, with much less resolution for blocks <50 kb as expected for small block sizes (STAR Methods S10a).  Figure 3 Mismatch Distributions by Block Size (A) Heatmap showing the mismatch distribution of high-confidence Denisovan introgressed blocks against the Altai Denisovan (x axis; expressed as a percentage of the average genome-wide mismatch between the Denisovan genome and West Eurasians, mD (STAR Methods S10a) given different minimum block lengths (y axis). The columns to the right indicate the total number of blocks and the number of entirely non-overlapping (i.e., unambiguously unique) blocks, respectively. As expected (see STAR Methods 10a for explanation), the two mismatch peaks are not visible for small block lengths (<50 kb), and only become clear for blocks of length around ≥130 kb. The two mismatch peaks are stable in their location after this point. (B) Heatmap as in (A), this time showing the mismatch distribution when analyzing Siberian and East Asian samples together. Note the absence of the first peak observed in Papuans in (A) and the presence of the less diverged peak identified previously (Browning et al., 2018). (C) Mismatch mD of the 2000 longest high-confidence Denisovan blocks for different continental groups. Circle area corresponds to the average amount per individual (Mb) of high-confidence Denisovan sequence identified in each population. We also confirm the signal previously reported in East Asians (Browning et al., 2018), but again only for longer block lengths (Figures 3B and 3C). The mismatch peak (here called D0) is additionally seen not just in East Asian populations, where it was originally detected, but also in Siberians, indigenous Americans and at very low frequency elsewhere across Asia (Figures 3B and 3C; STAR Methods S10a). The blocks in this predominantly East Asian mismatch peak have relatively low divergence to the Altai Denisovan, suggesting that modern humans in Asia mixed with a Denisovan population that was closely related to the Denisovan reference individual. Figure S2 Assessing the Mismatch Distribution in East ISEA and Resolution to Distinguish Mismatch Peaks Using Different Block Lengths, Related to Figure 3 and STAR Methods As longer blocks are better able to capture demographic complexity from mismatch distributions, we profiled mismatch patterns using the 2,000 longest blocks to maximize the signal in each population. Regional patterns are apparent: D1 is restricted to Papuans, while D2 has a wider geographical distribution spanning much of Asia and Oceania (Figure 3C; STAR Methods S10a; Figure S2).  Figure 4 Multiple Denisovan Ancestries in Papuans (A) Bimodal distribution and simulation fitting for long (>180 kb) high-confidence Denisovan blocks in Papuans (STAR Methods S10a and S10d). (B) Schematic model of the relationships of archaic hominin and modern human groups. We detect introgression from three Denisovan-like populations: the previously reported D0 lineage (gray) into East Asians (Browning et al., 2018) and Siberians with a likely recent introgression date, and the D1 (green) and D2 (magenta) lineages into Papuans that are detected here. The tree topologies of D1 and D2 haplotypes indicate that they branch from the Denisovan clade, albeit deeply for D2, and their bimodal mismatch indicates that they are not sister clades. Note the different timescales of the two orthogonal trees. Shaded circles on archaic tree branches indicate introgression time estimates. The details of these estimates (STAR Methods S10h) are shown on the modern human tree; the 95% CIs of introgression dates are shaded with color proportional to probability density, with bootstrapped values showing the center of gravity lies toward younger dates. No time estimates were reported for D0 in the original publication and its divergence from the Altai Denisovan and introgression date are arbitrarily placed as indicated by dashed lines. The open diamond indicates the temporal sampling point of the Altai Denisovan genome sequence. See also Figures S3, S4, and S5 and Table S5. Gaussian mixture model testing strongly supports the presence of two peaks in Papuans (Figure 4A, AIC = −5,809 versus unimodal −5,583, STAR Methods S10d). In Papuans, blocks of length >180 kb were assigned to one or the other peak based on >80% support from the mixture model. We confirmed that D1 (less divergent from Altai Denisovan) and D2 (more divergent) blocks do not differ in a wide range of molecular genetic and bioinformatic parameters, including GC content, genotype call quality of the archaic reference allele, alignability, recombination rate, sequencing batch effects, and levels of background selection (B values; McVicker et al., 2009) (STAR Methods 10e and 10f). We also checked whether variants within the D1 and D2 blocks have the same tree topologies. Both peaks show a strong signal of an origin within the Denisovan clade rather than branching from deeper points in the hominin tree (Table S4; STAR Methods S10g).  Figure S3 Mismatch Distributions Using Different Block Sets, Related to Figure 3 and STAR Methods We further verified that the observed bimodal mismatch distribution in our high-confidence Denisovan blocks is not due to misclassification of Neanderthal blocks. The polymorphic sites of both peaks predominantly show the Denisovan-specific topology, with the Neanderthal-specific topology observed only at low levels (STAR Methods S10g). Further, if one of the peaks were caused by Neanderthal introgression misclassified as Denisovan, that peak should be seen in West Eurasians, who have substantial Neanderthal but no Denisovan admixture. However, West Eurasians have neither of the two Denisovan mismatch peaks. To additionally check whether some portion of the Neanderthal introgression signal could have been missed by only using the Altai Neanderthal reference, we repeated several analyses using CP and the Vindija Neanderthal (Prüfer et al., 2017). This approach yields highly consistent results with the original analysis (Figure S3; STAR Methods S10c). Finally, we identified Neanderthal-specific blocks in Papuans using the same methodology as for the high-confidence Denisovan blocks. These do not show a bimodal mismatch distribution to the Altai Neanderthal (Figure S3; STAR Methods S10c), suggesting that the history of Denisovan introgression in Papua differed markedly from modern human interactions with Neanderthals. Next, we sought to retrieve dates of divergence between D1, D2, and the Altai Denisovan genome through coalescent modeling (Tables S5A and S5B; STAR Methods S10i). After extending an archaic demographic model (Malaspinas et al., 2016) to encompass two deeply divergent Denisovan-related components, our best fitting model indicates that D1 and D2 split from the Altai Denisovan approximately 283 kya (9,750 generations, 95% confidence interval [CI] 261–297 kya) and 363 kya (12,500 generations, 95% CI 334–377 kya), respectively (Figure 4B). While clearly branching off the Denisovan line, D2 diverged so closely to the Neanderthal-Denisovan split that it is perhaps better considered as a third sister group (STAR Methods S10i). For context, even the youngest of these divergence times is similar to the evolutionary age of anatomically modern humans (earliest known fossils, with varied morphologies, date to 198 kya (McDougall et al., 2005) and 315 kya (Hublin et al., 2017)). Our model implies substantial reproductive separation of multiple Denisovan-like populations over a period of hundreds of thousands of years. |
|
|
|
Post by Admin on Apr 13, 2019 17:51:51 GMT
Deep Divergence between Denisovan Populations Next, we sought to retrieve dates of divergence between D1, D2, and the Altai Denisovan genome through coalescent modeling (Tables S5A and S5B; STAR Methods S10i). After extending an archaic demographic model (Malaspinas et al., 2016) to encompass two deeply divergent Denisovan-related components, our best fitting model indicates that D1 and D2 split from the Altai Denisovan approximately 283 kya (9,750 generations, 95% confidence interval [CI] 261–297 kya) and 363 kya (12,500 generations, 95% CI 334–377 kya), respectively (Figure 4B). While clearly branching off the Denisovan line, D2 diverged so closely to the Neanderthal-Denisovan split that it is perhaps better considered as a third sister group (STAR Methods S10i). For context, even the youngest of these divergence times is similar to the evolutionary age of anatomically modern humans (earliest known fossils, with varied morphologies, date to 198 kya (McDougall et al., 2005) and 315 kya (Hublin et al., 2017)). Our model implies substantial reproductive separation of multiple Denisovan-like populations over a period of hundreds of thousands of years.  Figure S4 Using Simulations to Assess the Accuracy of Introgression Time Inference Based on Exponential Fitting of Block Length Distributions, and Example Fits, Related to Figures 4 and 5 and STAR Methods The Two Denisovan Lineages Introgressed at Different Times The distribution of block lengths retains a signal of introgression time, with longer blocks expected from more recent introgression events. In general, block length is expected to decay over time approximately as an exponential distribution (Gravel, 2012). We confirmed the accuracy of introgression dating by exponential fitting of the block length distribution through extensive simulation, incorporating different introgression times over the time period of interest (0–2,000 generations), and considering the impact of using only long blocks rather than the entire distribution of block lengths, substantial block length estimation errors, and the consequences if introgression occurred as an extended process rather than a single pulse (Figure S4; STAR Methods S10h). We observed a slight tendency to infer overly recent dates under some of these conditions, but never by more than 10%–15%. Filtering to longer block lengths and fitting an exponential with a larger location parameter help to reduce even these biases in date estimates. While the median block lengths of D1 and D2 are similar in Papuans (238 and 236 kb), their distributions are significantly different (Kolmogorov-Smirnov statistic = 0.15, p = 2.2 × 10−6). Exponential fitting of D1 and D2 haplotype lengths yields introgression dates of 29.8 kya (95% CI 14.4–50.4) and 45.7 kya (95% CI 31.9–60.7), respectively, which are younger, though overlapping with, previously suggested estimates for Denisovan introgression (Figure 4B; STAR Methods S10h) (Malaspinas et al., 2016). The maximum likelihood introgression date for D2 introgression is 50% more ancient than the date for D1. Based on simulations, and given the greater statistical challenge of identifying shorter introgression blocks, we consider these dates to be probable lower bounds on introgression times, but with true dates no more than 15% more ancient. Geographical Patterns of Denisovan Admixture in Papua D1 and D2 introgression times that overlap the timescale of modern human arrival and their variable dispersal across Papua raise the possibility that Denisovan introgression occurred after local populations of modern humans had differentiated. We find geographic structure associated with the D1 variation between mainland New Guinea and the Baining, a population on the offshore island of New Britain. We observe slightly less high-confidence Denisovan introgression in the Baining than in mainland Papuans (31.5 Mb versus 33.1 Mb per haploid genome, Welch’s t test T = −3.4, p = 0.001), despite extremely similar population histories (Hudjashov et al., 2017), including similar levels of Asian ancestry (Figure 1A). However, there is less D1 sequence in the Baining than in mainland Papuans (1.33 Mb versus 1.82 Mb per haploid genome, Welch’s t test T = −3.9, p < 0.01), although both carry similar levels of D2 sequence (1.28 Mb versus 1.37 Mb, T = −0.8, p = 0.41) (Figures 5A and 5B; STAR Methods S10h).  Figure 5 Geographic Patterns of D1 and D2 Ancestry To determine whether this difference in D1 sequence could be due to random drift in the two populations or to different Denisovan introgression histories, we extended the simulation model (Malaspinas et al., 2016) to incorporate population structure representing both New Guinea mainlanders and Baining, in addition to the two introgressing Denisovan populations (D1 and D2) (Figure S5; STAR Methods S10j). To test a conservative model offering maximum opportunity for isolation and drift, we did not include any migration between Papuans and Baining after their population split. Archeological evidence suggests that New Britain was settled by at least 35 kya (Pavlides and Gosden, 1994), and from the genomic data, SMC++ (Terhorst et al., 2017) infers a genetic split time between mainland Papuans and Baining of 15.7 kya (Figure 5C). We therefore implemented three alternative demographic models: using the SMC++ genetic split times and population sizes (M1); using the SMC++ split time and more conservative (smaller) population sizes, thus generating more drift (M2); and a model with a more conservative (older) genetic split time of 23.2 ky (800 generations), also generating more drift (M3) (Figure S6; STAR Methods S10j). As expected, the observed difference in rates of D2 introgression between Baining and mainland Papuans are within the distributions predicted by the simulations. However, in all three cases, the observed ratio of D1 in mainland Papuans to Baining lies outside simulated values (Figure 5D).  Figure S5 Simulation Model Schematic and Mismatch Results, Related to Figure 4 and STAR Methods Together, these coalescent simulations suggest that the reduced frequency of D1 blocks among the Baining is unlikely to result from shared D1 introgression into a common ancestral Papuan population, followed by drift as each population subsequently diverged into the modern Baining and mainland groups. Instead, the difference in D1 levels more likely reflects different amounts of introgression from Denisovan populations into mainland New Guinea and the islands to the northeast, which occurred after the separation of the two Papuan populations (Figure 5E). The overall genetic similarity and relatively recent divergence of these Papuan groups (Figures 1 and 5C; STAR Methods S10h, S10j) have implications for the past distribution of D1 Denisovan populations and the process of archaic introgression. First, our data suggest that the D1 Denisovans, in contrast to D2, contributed additional DNA to the mainland New Guinea population after the mainland and Baining populations diverged from their common Papuan ancestor (Figure 5E). This, together with the nearly complete absence of D1 in continental Asia, is most consistent with the scenario that D1 Denisovans were present in New Guinea or East ISEA (e.g., Wallacea). In turn, this would imply that at least some Denisovan populations had the ability to cross large bodies of water, such as the one represented by the Wallace Line. This idea does not seem implausible given archaeological evidence of archaic hominin dispersals—notably, the discovery of stone tools in the Philippines dating to 700 kya (Ingicco et al., 2018) and the related finding of H. floresiensis on the island of Flores (Brown et al., 2004), both across substantial water boundaries that persisted throughout the Pleistocene. Such geographical barriers would limit gene flow and might help to explain the extent of divergence between the D1 Denisovan population and other Denisovan groups.  Figure S6 Using Heterozygosity and Fst to Inform Models of Mainland Papuan/Baining Drift, Related to Figure 5 and STAR Methods Second, the late date for the D1 introgression and geographic structure in modern populations suggests that Denisovans survived until 30 kya, and perhaps as recently as 14.5 kya. This is longer than Neanderthals, who died out around 40 kya (Higham et al., 2014), or H. floresiensis, which recent dating suggests did not persist on Flores beyond 50–60 kya (Sutikna et al., 2016). The implication is that Denisovans living in ISEA may have been among the last of all the archaic hominins to survive. This provides an argument to screen for Denisovan remains possibly misclassified as other hominins in existing archaeological collections and encourages more archaeological research in the poorly accessible and hence incredibly understudied New Guinea region. Third, the combined evidence of geographic structure and a recent D1 introgression date suggest that Denisovan introgression did not occur immediately following the first modern human settlement in the region (45–50 kya) (O’Connell et al., 2018). This implies that introgression with archaic hominins may not be an inevitable and immediate result of joint occupation of the same territory. |
|

