|
|
Post by Admin on Sept 9, 2023 20:38:09 GMT
Specific changes in DNA may increase the risk of developing epilepsy, according to research published in the journal Nature GeneticsTrusted Source. In the study, researchers identified 26 areas of DNA involved in the development of epilepsy and 29 genes that are likely contributing to epilepsy within those areas. Dr. Gianpiero Cavalleri, PhD, a co-author of the study and a professor of human genetics at RCSI School of Pharmacy and Biomolecular Science and Deputy Director of the SFI FutureNeuro Research Centre, said in a press statement: “Gaining a better understanding of the genetic underpinnings of epilepsy is key to developing new therapeutic options and consequently a better quality of life for the over 50 million peopleTrusted Source globally living with epilepsy. The discoveries we report on here could only be achieved through international collaboration, on a global scale. We are proud of how the global community of scientists working to better understand the genetics of the epilepsies have pooled resources and collaborated effectively, for the benefit of people impacted the condition.” Largest genetic study to identify DNA changes associated with epilepsy The new research is the largest genetic study of its kind and involved more than 150 researchers in North America, South America, Europe, Australia and Asia. As part of the study, the researchers compared DNA from nearly 30,000 people living with epilepsy alongside DNA from more than 52,500 people without the condition. The scientists were able to identify 26 distinct changes to the DNA in people living with epilepsy. They were also able to identify 19 changes in DNA that are specific to a type of epilepsy called genetic generalized epilepsy (GGE). “This identification of epilepsy associated genetic changes will allow us to improve diagnosis and classification of different epilepsy subtypes. This in turn, will guide clinicians in selecting the most beneficial treatment strategies, minimising seizures” Dr. Colin Doherty, a co-author of the study as well as a consultant neurologist at St James’s Hospital in Dublin and clinical investigator at the SFI FutureNeuro Centre, said in a press statement. As part of the study, the researchers said they were able to demonstrate that many medications currently used in the treatment of epilepsy work by targeting the genes they identified as increasing the risk of epilepsy. They were also able to use their data to suggest alternative drugs that are currently used for other conditions but are also known to target some of the genes identified in the study. Understanding the genetics of epilepsy could lead to improved therapies Epilepsy is a brain disorder in which neurons, a type of nerve cell located in the brain, can sometimes send the wrong signals. This causes seizures. When a seizure occurs, there is an increase in excessive electrical activity in the brain. This may cause involuntary sensations, movements and behavior. Experts say the study represents an important step forward in understanding epilepsy. “We don’t really understand clearly what are the causes of epilepsy and how it comes about,” Dr. Jean-Philippe Langevin, a neurosurgeon and director of Restorative Neurosurgery and Deep Brain Stimulation Program for Pacific Neuroscience Institute at Providence Saint John’s Health Center in California, told MNT. “This study goes a long way to provide some understanding, like the biology behind the formation of epilepsy. Those genetic differences leads the researcher to postulate about some proteins and some molecules that might be involved in epilepsy in all these patients. The more we understand about the genetics, it’s also going to provide new targets for future therapy. The more we understand the biology and the physiology of the disease, the better we can target and offer treatments for the patients.” — Dr. Jean-Philippe Langevin, neurosurgeon Dr. Clifford Segil, a neurologist at Providence Saint John’s Health Center in California, told MNT that experts still don’t fully understand what exactly makes a seizure start or stop. “Our knowledge, when it comes to neuroscience, is still very basic and benign,” Dr. Segil said. “If we have an understanding of the genetics of epilepsy, it will help patients who have seizures get better medical treatments. “The mainstay of treating patients with seizures is to give them anti-seizure medications, or anti-epileptic drugs. The drugs, by definition make your brain electricity fire slower because a seizure is an uncontrollable burst of electricity. And these medications can cause people to be slower fatigued. With a genetic understanding of seizures, we would have a molecular basis of what is causing the seizure. So a gene test will help an epileptologist determine which drugs to use in a perfect world.” — Dr. Clifford Segil, neurologist www.nature.com/articles/s41588-023-01485-w |
|
|
|
Post by Admin on Sept 10, 2023 21:58:30 GMT
GWAS meta-analysis of over 29,000 people with epilepsy identifies 26 risk loci and subtype-specific genetic architecture
Abstract
Epilepsy is a highly heritable disorder affecting over 50 million people worldwide, of which about one-third are resistant to current treatments. Here we report a multi-ancestry genome-wide association study including 29,944 cases, stratified into three broad categories and seven subtypes of epilepsy, and 52,538 controls. We identify 26 genome-wide significant loci, 19 of which are specific to genetic generalized epilepsy (GGE). We implicate 29 likely causal genes underlying these 26 loci. SNP-based heritability analyses show that common variants explain between 39.6% and 90% of genetic risk for GGE and its subtypes. Subtype analysis revealed markedly different genetic architectures between focal and generalized epilepsies. Gene-set analyses of GGE signals implicate synaptic processes in both excitatory and inhibitory neurons in the brain. Prioritized candidate genes overlap with monogenic epilepsy genes and with targets of current antiseizure medications. Finally, we leverage our results to identify alternate drugs with predicted efficacy if repurposed for epilepsy treatment.
Main
The epilepsies are a heterogeneous group of neurological disorders, characterized by an enduring predisposition to generate unprovoked seizures1. It is estimated that over 50 million people worldwide have active epilepsy, with an annual cumulative incidence of 68 per 100,000 persons2.
Similar to other common neurodevelopmental disorders, epilepsies have substantial genetic risk contributions from both common and rare genetic variations. Analysis of the epilepsies benefits from deep phenotyping, which allows clinical subtypes to be distinguished3, in contrast to other common neurodevelopmental disorders, where phenotypic subtypes are more difficult to define. Differences in the genetic architecture of clinical subtypes of epilepsy are also emerging, to complement the clinical partitioning4,5,6,7. The rare but severe epileptic encephalopathies are usually nonfamilial and are largely caused by single de novo dominant variants, often involving genes encoding ion channels or proteins of the synaptic machinery8. Both common and rare variants have been shown to contribute to the milder and more common focal and generalized epilepsies. This is particularly true for generalized epilepsy, which is primarily constituted by genetic generalized epilepsy (GGE)4,5,9,10. Nevertheless, previous genetic studies of common epilepsies have explained only a limited proportion of this common genetic variant, or single-nucleotide polymorphism (SNP)-based, heritability—9.2% for focal and 32.1% for GGE4,5,6,10.
Epilepsy is typically treated using antiseizure medications (ASMs). However, despite the availability of over 25 licensed ASMs worldwide, a third of people with epilepsy experience continuing seizures11. Diet, surgery and neuromodulation represent additional treatment options that can be effective in small subgroups of patients12. Accurate classification of clinical presentations is an important guiding factor in epilepsy treatment.
Here we report the third epilepsy genome-wide association study (GWAS) meta-analysis by the International League against Epilepsy (ILAE) Consortium on complex epilepsies, comprising a total of 29,944 deeply phenotyped cases recruited from tertiary referral centers and 52,538 controls, approximately doubling the previous sample size4. Results suggest markedly different genetic architectures between focal and generalized forms of epilepsy. Combining these results with those from less-stringently phenotyped biobank and deCODE genetics epilepsy cases did not substantially increase signal, despite almost doubling the sample size to 51,678 cases and 1,076,527 controls. Our findings shed light on the enigmatic biology of generalized epilepsy and the importance of accurate syndromic phenotyping and may facilitate drug repurposing for new therapeutic approaches.
|
|
|
|
Post by Admin on Sept 12, 2023 20:37:28 GMT
Results Study overview We performed a GWAS meta-analysis by combining the previously published effort from our consortium4 with unpublished data from the Epi25 collaborative10 and four additional cohorts (Supplementary Tables 1 and 2). Our primary mixed model meta-analysis constitutes 4.9 million SNPs tested in 52,538 controls and 29,944 people with epilepsy, of which 16,384 had neurologist-classified focal epilepsy (FE) and 7,407 had GGE. The epilepsy cases were primarily of European descent (92%), with a smaller proportion of African (3%) and Asian (5%) ancestry (Supplementary Table 3). Cases were matched with controls of the same ancestry, and GWAS analyses were performed separately per ancestry, before performing multi-ancestry meta-analyses for the broad epilepsy phenotypes ‘FE’ (n = 16,384 cases) and ‘GGE’ (n = 7,407 cases). We further conducted meta-analyses in individuals of European ancestry of the well-defined GGE subtypes of juvenile myoclonic epilepsy (JME; n = 1,732), childhood absence epilepsy (CAE; n = 1,049), juvenile absence epilepsy (JAE; n = 662) and generalized tonic-clonic seizures alone (GTCSA; n = 485), as well as the FE subtypes of FE with hippocampal sclerosis (HS; n = 1,260), FE with other lesions (n = 4,213) and lesion-negative FE (n = 5,778). The same controls (n = 42,436) were shared across the different subphenotypes. We ran a variety of follow-up analyses to identify potential sex-specific signals and obtain biological insights and opportunities for drug repurposing. Sample size prevented the inclusion of other ethnicities in the subtype analyses. GWAS for the epilepsies Our ‘all epilepsy’ meta-analysis revealed four genome-wide significant loci, of which two are new (Fig. 1). Similar to our previous GWAS4, the 2q24.3 locus was composed of two independently significant signals (Supplementary Table 4). Using ASSET to determine the extent of FE and GGE-related pleiotropy, the 2q24.3 and 9q21.13 signals showed pleiotropic effects at a genome-wide significance level, with concordant SNP effect directions for both forms of epilepsy (Supplementary Table 5). The 2p16.1 and 10q24.32 loci were primarily derived from GGE. The FE analysis did not reveal any genome-wide significant signals. Fig. 1: Manhattan plot of multi-ancestry all epilepsy (n = 29,944), focal epilepsy (n = 16,384) and genetic generalized epilepsy (n = 7,407) genome-wide meta-analyses, obtained by fixed-effects meta-analysis weighted by effective sample sizes. 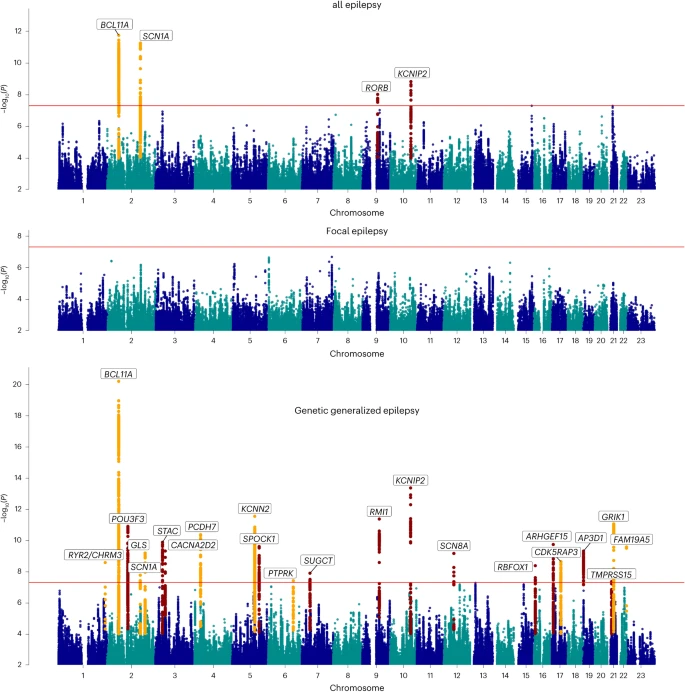 The red line shows the genome-wide significance threshold (5 × 10−8). Chromosome and position are displayed on the x axis, and two-sided −log10 P value is on the y axis. New genome-wide significant loci are highlighted in red, and loci previously associated with epilepsy in orange. New loci were those previously unreported as GWAS significant in previous epilepsy GWASs. Annotated genes are those implicated by our gene prioritization analyses. See Supplementary Fig. 7 for QQ plots. QQ plots, quantile–quantile plot. Our ‘GGE’ meta-analysis uncovered a total of 25 independent genome-wide significant signals across 22 loci, of which 13 loci are new. The strongest signal of association (P = 6.6 × 10−21), located at 2p16.1, constitutes three independently significant signals. Similarly, the new locus 12q13.13 was composed of two independently significant signals (Supplementary Table 4). Forest plots and P–M plots of these signals show that they appear consistent across all four GGE subphenotypes, with some exceptions (Supplementary Figs. 1 and 2). We applied multitrait analysis of GWAS (MTAG)17 to exploit the correlation between FE and GGE, boosting the effective sample size. Results were concordant with our main analysis, and new signals did not emerge (Supplementary Fig. 3). Functional annotation of the 1,082 genome-wide significant SNPs across the 22 GGE loci and 270 SNPs from the ‘all epilepsy’ loci revealed that most variants were intergenic or intronic (Supplementary Data 1). Eight of 1,082 (0.7%) GGE SNPs were exonic, of which five were located in protein-coding genes and were missense variants. We identified one exonic ‘all epilepsy’ SNP (rs7580482, synonymous), located in SCN1A. Seventy-four percent of ‘all epilepsy’ SNPs and 64% of GGE SNPs were located in open chromatin regions, as indicated by a minimum chromatin state of 1–7 (ref. 14). Further annotation by Combined Annotation-Dependent Depletion (CADD) scores predicted that 11 ‘all epilepsy’ and 50 GGE SNPs were deleterious (CADD score > 12.37) (ref. 15). LDAK heritability analyses showed significant enrichment of signal in ‘super-enhancers’ (Supplementary Table 6), suggesting that GGE SNPs regulate clusters of transcriptional enhancers that control the expression of genes that define cell identity16. To assess potential syndrome-specific loci, we performed GWAS on seven well-defined FE and GGE subtypes (Supplementary Fig. 4a–g). We found three genome-wide significant loci associated specifically with JME (n = 1,813), of which one was new (8q23.1) and the other two (4p12 and 16p11.2) previously reported4. Our analysis of CAE (n = 1,072) consolidated an established genome-wide significant signal at 2p16.1, which was also observed in the GGE and all epilepsy GWAS. We did not find any genome-wide significant loci for JAE (n = 671), GTCSA (n = 499), ‘nonlesional FE’ (n = 6,367), ‘FE with HS’ (n = 1,375) or ‘FE with other lesions’ (n = 4,661). MTAG17 analysis of individual GGE subphenotypes showed concordance with the main GGE GWAS, without identifying new loci. In addition, this analysis confirmed that the majority of GWAS-significant SNPs in GGE are overlapping (Supplementary Figs. 5 and 6 and Supplementary Table 7). The vast majority of loci reported in our previous effort4 remained genome-wide significant. A summary of loci that fell below the genome-wide significance threshold is provided in Supplementary Table 8. Genomic inflation was comparable to our previous GWAS, and all linkage-disequilibrium score regression (LDSC) intercepts were lower (Supplementary Table 9)4, suggesting that the signals are primarily driven by polygenicity. Computation of the attenuation ratio suggested that part of the inflation signal, in particular for FE (0.58), might be due to some form of bias (for example, confounding or population stratification)13. The attenuation ratio was lowest for GGE (0.11), which includes the vast majority of significant loci (Supplementary Table 9). |
|
|
|
Post by Admin on Sept 14, 2023 21:51:49 GMT
Locus annotation, gene-based analyses and gene prioritization
Using FUMA18 (Methods), the ‘all epilepsy’ meta-analysis was mapped to 43 genes and the GGE analysis to 278 genes (Supplementary Data 2). Thirty-nine of the 43 ‘all epilepsy’ genes overlapped with GGE, resulting in a total of 282 uniquely mapped genes. These 282 genes were enriched for monogenic epilepsy genes (hypergeometric test, 18/837 genes overlapped; odds ratio (OR) = 1.51, P = 0.04) and targets of ASMs (hypergeometric test, 9/191 genes overlap; OR = 3.39, P = 5.4 × 10−4).
We calculated a gene-based association score based on the aggregate of all SNPs inside each gene using MAGMA (Methods)19. This analysis yielded 39 significant genic associations—six with ‘all epilepsy’ and 37 with GGE (four overlapped with the ‘all epilepsy’ analysis), after correction for 16,371 tested genes (P < 0.05/16,371 genes; Supplementary Data 3). Thirteen of these 39 genes mapped to regions outside of the genome-wide significant loci from the single SNP analyses.
Next, we performed a transcriptome-wide association study (TWAS) to assess whether epilepsy was associated with differential gene expression in the brain (Methods)20,21. These analyses revealed significant associations with 27 genes in total; 13 genes with ‘all epilepsy,’ 16 with GGE and two with both phenotypes (Supplementary Data 4). Nineteen of the 27 genes mapped outside of the 26 loci were identified through the GWAS. Using summary-data-based Mendelian randomization (SMR)22, we determined a potentially causal relationship between brain expression of RMI1 and ‘all epilepsy,’ and among RMI1, CDK5RAP3 and TVP23B and GGE (Supplementary Data 5).
Of note, expression of RMI1 was associated with GGE in both TWAS (P = 4.0 × 10−10) and SMR (P = 5.2 × 10−8), as well as with ‘all epilepsy’ (TWAS P = 1.3 × 10−6; SMR P = 2.6 × 10−6). RMI1 has a crucial role in genomic stability23 and has not been previously associated with epilepsy or any other Mendelian trait (OMIM, 610404).
We used a combination of ten different criteria to identify the most likely implicated gene within each of the 26 associated loci from the meta-analysis (Methods). This resulted in a shortlist of 29 genes (Table 1; see Supplementary Data 6 for scores of all mapped genes), of which ten are monogenic epilepsy genes, seven are known targets of currently licensed ASDs and 17 are associated with epilepsy for the first time.
The strongest association signal for GGE was found at 2p16.1, consistent with our previous results where we implicated VRK2 or FANCL24. Our gene prioritization analysis suggests the transcription factor BCL11A as the culprit gene, located 2.5 Mb upstream of the lead SNPs at this locus. Two of three lead SNPs are in enhancer regions (as assessed by chromatin states in brain tissue) that are linked to the BCL11A promoter via 3D chromatin interactions (Supplementary Fig. 8). Rare variants in BCL11A were recently associated with intellectual disability and epileptic encephalopathy25. However, interrogation of the MetaBrain expression quantitative trait loci (eQTL) database did not reveal a significant association of our lead SNPs with BCL11A expression.
|
|
|
|
Post by Admin on Sept 16, 2023 21:46:00 GMT
The HLA system and common epilepsies
The highly polymorphic HLA region has been associated with various neuropsychiatric and autoimmune neurological disorders. Therefore, we imputed HLA alleles and amino acid residues using CookHLA v1.0.1 (ref. 26) and ran association across epilepsy, focal and GGE phenotypes, as well as the seven subphenotypes (Methods). No SNP, amino acid residue or HLA allele reached genome-wide significance (Supplementary Fig. 9). The most significant signal was an aspartame amino acid residue in exon 2 of HLA-B (position 31432494), which had a P value of 3.8 × 10−7 for GGE.
SNP-based heritability
We calculated SNP-based heritability using LDAK to determine the proportion of epilepsy risk attributable to common genetic variants. We observed liability scale SNP-based heritabilities of 17.7% (95% confidence interval (CI): 15.5–19.9%) for all epilepsy, 16.0% (14.0–18.0%) for FE and 39.6% (34.3–44.6%) for GGE. Heritabilities were notably higher for all individual GGE subtypes, ranging from 49.6% (14.0–85.3%) for GTCSA to 90.0% (63.3–116.6%) for JAE (Supplementary Table 10).
Using a univariate causal mixture model27 (Methods), we estimated that 2,850 causal SNPs (s.e.: 200) underlie 90% of the SNP-based heritability of GGE, comparable with previous estimates9. Power analysis demonstrated that the current genome-wide significant SNPs only explain 1.5% of the phenotypic variance, whereas an estimated sample size of around 2.5 million individuals would be necessary to identify the causal SNPs that explain 90% of GGE SNP-based heritability (Supplementary Fig. 10).
To further explore the heritability of the different epilepsy phenotypes, we used LDSC to perform genetic correlation analyses28. We found evidence for a strong genetic correlation among all four GGE syndromes (Supplementary Fig. 11 and Supplementary Table 11). We also observed the previously reported significant genetic correlation4 between the focal nonlesional and JME syndromes. Here CAE also showed a significant genetic correlation with the focal nonlesional cohort. Multivariate modeling of genetic correlation using Genomic structural equation modeling (SEM)29 confirmed that most of the heritability signal is shared among the four GGE syndromes, with some subtype-specific signals (Supplementary Fig. 12).
Tissue and cell type enrichment
To further illuminate the underlying biological causes of the epilepsies, we used MAGMA19 and data from the gene–tissue expression (GTEx) consortium to assess whether our GGE-associated genes were enriched for expression in specific tissues and cell types (Methods). We identified significant enrichment of associated genes expressed in brain and pituitary tissue (Supplementary Fig. 13). The implication of the pituitary gland in GGE might reflect a hormonal component to seizure susceptibility. Further subanalyses showed that our results were enriched for genes expressed in almost all brain regions, including subcortical structures such as the hypothalamus, hippocampus and amygdala (Supplementary Fig. 14). We did not find enrichment for genes expressed at specific developmental stages in the brain (Supplementary Fig. 15).
Cell-type specificity analyses of GGE data using various single-cell RNA-sequencing reference datasets (Methods) revealed enrichment in excitatory as well as inhibitory neurons, but not in other brain cells like astrocytes, oligodendrocytes or microglia (Supplementary Fig. 16). Similarly, stratified linkage-disequilibrium (LD)-score regression using single-cell expression data (Methods) did not reveal a difference between excitatory and inhibitory neurons (P = 0.18).
|
|

