|
|
Post by Admin on Sept 18, 2016 21:30:00 GMT
 Millions of men bear the genetic legacy of Genghis Khan, the famously fertile Mongolian ruler who died in 1227. Researchers have now recognized ten other men whose fecundity has left a lasting impression on present-day populations. The team's study1 points to sociopolitical factors that foster such lineages, but the identities of the men who left their genetic stamp remains unknown. The case for Genghis Khan’s genetic legacy is strong, if circumstantial. A 2003 paper2 led by Chris Tyler-Smith, an evolutionary geneticist now at the Wellcome Trust Sanger Institute in Hinxton, UK, discovered that 8% of men in 16 populations spanning Asia (and 0.5% of men worldwide) shared nearly identical Y-chromosome sequences. The variation that did exist in their DNA suggested that the lineage began around 1,000 years ago in Mongolia.  Genghis Khan is reputed to have sired hundreds of children. But a Y-chromosome lineage traces a single paternal line in a much larger family tree, and for it to leave a lasting legacy takes multiple generations who fan out over a wide geographical area, says Mark Jobling, a geneticist at the University of Leicester, UK, who led the latest study with geneticist Patricia Balaresque of Paul Sabatier University in Toulouse, France. Jobling’s team made a systematic search for genetic founders by analysing the Y chromosomes of more than 5,000 men from 127 populations spanning Asia; they focused on that region because lots of data were available and there was already evidence of such lineages. The team identified 11 Y-chromosome sequences that were each shared by more than 20 of the 5,321 genomes. The researchers used DNA differences in the shared sequences, which accumulate over time from random mutations, to determine approximately when the founder of the lineage lived. They tracked back the geographical origins of the lineages by assuming that the founding men had lived in the regions where their genotypes were most prevalent and diverse.  Genghis Khan’s paternal lineage again stood out, as did Giocangga’s, Jobling’s team reports in the European Journal of Human Genetics1. The other nine lineages originated throughout Asia, from the Middle East to southeast Asia, dating to between 2100 bc and ad 700. Jobling warns that these dates come with huge margins of error, but he notes that the estimates for the lineages attributed to Khan and Giocangga are very close to those of past studies.  Legacy of power The founders who lived between 2100 bc and 300 bc existed in both sedentary agricultural societies and nomadic cultures in the Middle East, India, southeast Asia and central Asia. Their dates coincide with the emergence of hierarchical, authoritarian societies in Asia during the Bronze Age, such as the Babylonians. Three lineages dating to more recent times were all linked to nomadic groups in northeast China and Mongolia. These included the lineages linked to Genghis Khan and Giocangga, plus a third line dating to around ad 850. All three lineages seem to have expanded westwards, possibly along the Silk Road trade route. Historians have documented a series of polities based in inner Asia between 200 bc and the eighteenth century, such as the Qing Dynasty. Jobling says that these civilizations could have fostered dominant male lineages after the sons of a fecund founder decamped to satellite outposts, where they, in turn, fathered powerful descendants. |
|
|
|
Post by Admin on Sept 19, 2016 20:59:39 GMT
 Fig 1. Geographical features of Tavan Tolgoi graves. 7 graves were first excavated in the central hill of Tavan Tolgoi (“five hills” in the Mongolian language) by a Mongolian excavation team. Tavan Tolgoi lies within the Ongud province once dominated by Alaqai Beki and then Sorkhokhtani, a wife of Genghis Khan’s youngest son Tolui, during the early Mongolian era [8]. Mongolian archaeologists who participated in the excavation of Tavan Tolgoi graves strongly suspected that 5 of 7 Tavan Tolgoi graves belonged to the Golden family [9–11], and one of those 5 graves was thought to be that of a Mongol Queen. Burial artifacts excavated from the Tavan Tolgoi graves have since been displayed in the National Museum of Mongolian History, recognized as important relics of the Mongol Empire. In this study, we aimed to determine the matrilineal and patrilineal origins of the Golden family members from the Tavan Tolgoi burial site and to determine kinship among them and with Genghis Khan, through analysis of single nucleotide polymorphism (SNP) and short tandem repeat (STR) of mitochondria, autosomes, and Y chromosomes from ancient DNAs (aDNAs). In addition, we compared their mitochondrial DNA (mtDNA) and Y-STR haplotypes with those of modern-day individuals using neighbor-joining (NJ) and Y-chromosome STR Haplotype Reference Database (YHRD) analyses, respectively, to determine the current geographical distribution of female- and male-line descendants of the Golden family members. Moreover, the familial relationship of the Tavan Tolgoi bodies with Genghis Khan was also postulated based on molecular archaeological and historical evidence. This study, the first molecular archaeological analysis of several skeletons belonging to Genghis Khan’s Golden family, presents molecular data to reveal the identity and genealogy of Golden family members, including a Mongol Queen, thereby unlocking the door to mysterious lives of the Golden family.  Fig 2. Some of the burial artifacts excavated from the Tavan Tolgoi graves. Golden family members of Tavan Tolgoi reveal the genealogical admixture between Caucasoid and Mongoloid ethnic groups So far, no molecular archaeological study of members of the Mongolian imperial family has been conducted; this is largely because no grave of imperial family, especially those of the Golden family, has been identified until the Tavan Tolgoi grave excavation. To the best of our knowledge, this study is the first molecular archeological attempt to define the genealogy of Genghis Khan’s Golden family members in the Mongolian era. Evidence suggests that many Mongoloid and Caucasoid nomadic tribes inhabited the present-day Mongolian plateau over thousands of years [40]. Since Genghis Khan’s era, the Mongolian population underwent rapid and considerable gene flow from Eurasia, resulting in additional genetic admixture [40]. Likewise, the Mongolian population was formed by the continuous admixture of indigenous tribes who inhabited the Mongolian plateau, with European and other Asian populations who inhabited regions geographically distant from Mongolia. This admixture has deeply influenced the physical appearance of modern-day Mongolian people, exhibiting both Mongoloid and Caucasoid features. 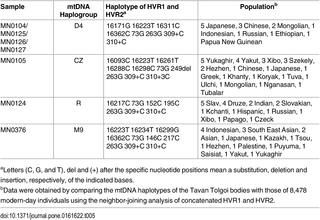 Table 5. Geographic distribution of the modern-day individuals with the same mtDNA haplotypes as those of the Tavan Tolgoi bodies. The mixing between Mongoloid and Caucasoid ethnic groups inherent in the genetic structure of modern-day Mongolians was also observed in the Tavan Tolgoi bodies. The Golden family members carried mtDNA haplogroups D4 and CZ, mostly found in Far Eastern and Northeastern Asia, respectively, whereas male members of Golden family carried the Y-haplogroup R1b-M343, dominant in Western Europe [41–43]. That is, although members of Golden family were physically Mongoloid, their molecular genealogy revealed the admixture between Caucasoid and Mongoloid ethnic groups. Thus, it is likely that their Mongoloid appearance would have resulted from gradual changes in their appearance from Caucasoid to Mongoloid through generations from their ancestors. Their physical appearance was largely attributed to D4-carrying Mongoloid females who were indigenous peoples of the Mongolian plateau, rather than an R1b-M343-carrying Caucasoid male spouse who had initially moved from Europe to the Mongolian plateau and his male descendants; it is, however, uncertain how and when the admixture between Mongoloid and Caucasoid ethnic groups originated in the Mongolian plateau. Y-haplogroup R1b-M343 of Tavan Tolgoi bodies may be another candidate for the Y-haplogroup of Genghis Khan and Genghis Khan’s Borjigin clan Although many regard the portrait at the National Palace Museum in Taipei, Taiwan, as the depiction most closely resembles Genghis Khan, all existing portraits, including this one, are essentially arbitrary interpretations of Genghis Khan’s appearance by historians living generations after Genghis Khan’s era [2, 6]. Although the factual nature of the statement is controversial, Persian historian Rashid-al-Din reported in his “Jami’s al-tawarikh” written at the start of the 14th century that most Borjigin ancestors of Genghis Khan were tall, long-bearded, red-haired, and bluish green-eyed, suggesting that the Genghis Khan’s male lineage had some Caucasoid-specific genetic features [44]. He also said that Genghis Khan looked just like his ancestors, but Kublai Khan, his grandson, did not inherit his ancestor’s red hair, implying that the addition of Mongoloid-specific alleles for determining hair color to the genetic makeup of Genghis Khan’s Borjigin clan was probably from the grandmother or mother of Kublai Khan, that is, the wife or daughter-in-law of Genghis Khan.  Fig 3. Geographic distribution of modern-day populations with haplogroup R1b-M343. “On” and “gud” from Ongud mean West and plural in ancient Altaic language, respectively, implying that the Ongud is a tribe from Western Asia. In fact, the ancestors of the Ongud are the Shato Turks of the Western Göktürks Khaganate [45, 46]; they moved to Eastern Xinjiang in the 7th century, and were scattered over Northern China and Inner Mongolia in the 9th century [47]. In the Mongolian era, many Ongud peoples were resettled in Khorazm of Western Central Asia, as governors for the Golden Horde Dynasty, and eventually formed part of the Kazakhs and the Mughals [44]. In addition, they also fell under the Chagatai Khanate that was ruled by Chagatai Khan and his descendants and/or successors and extended from the Southern part of the Aral Sea to the Altai Mountains [48]. These suggest the possibility that the Ongud clan may be anthropologically Caucasoid rather than Mongoloid, according to their geographical origin. Therefore, the male bodies carrying R1b-M343 (prevalent in Western Europe) from Tavan Tolgoi, which was located within the territory of the Ongud Kingdom during the early Mongolian era, could be related to the Ongud male lineage, implying that Tavan Tolgoi bodies are genealogically Caucasoid. Eastern Russian Tatars, Bashkirs, and Pakistani Hazara were found to carry R1b-M343 at unusually high frequencies of 12.65%, 46.07%, and 32%, respectively, compared to other regions of Eastern Asia, which rarely have this haplotype (Fig 3) [40, 42, 43, 49–53]. Interestingly, ancestors of those 3 populations were all closely associated with the medieval Mongol Empire. That is, Russian Tatars and Bashkirs are descendants of the Golden Horde (also known as the Ulus of Jochi) that had been controlled by Jochi, the first son of Genghis Khan, and his descendants during the 12th–15th centuries. In addition, some of the Hazara tribes are believed to consist of descendants of Mongolian soldiers and their slave women after the 1221 siege of Bamiyan under the leadership of Genghis Khan [54, 55]. Through domination of Hazara, Mongolians strongly influenced the genetic makeup of the Hazara people, especially in Pakistan [49, 54, 56]. Some modern Hazara populations resemble Mongolians in their physical attributes including facial bone structure. Similarly, the high frequency of R1b-M343 in geographic regions associated with the past Mongol khanates including the Golden Horde (from Ural Mountain to Western Siberia, which includes Russia, Ukraine, Belarus, Poland, Azerbaijan, Kazakhstan, and Uzbekistan), Ilkhanate (Iran and neighboring territories including Armenia, Turkey, Georgia, Afghanistan, Syria, and Tajikistan), and Chagatai Khanate (from the Aral sea to the Altai mountain, including Pakistan (Hazara), Uzbekistan, Kazakhstan, Tajikistan, India, and China), strongly suggest a close association between the Y haplotype R1b-M343 and the past Mongol Empire (Fig 3) [42–44, 49–53]. Lkhagvasuren G, Shin H, Lee SE, Tumen D, Kim J-H, Kim K-Y, et al. (2016) Molecular Genealogy of a Mongol Queen’s Family and Her Possible Kinship with Genghis Khan. PLoS ONE 11(9): e0161622. |
|
|
|
Post by Admin on Sept 21, 2016 21:00:19 GMT
 Figure 1. DNA samples (461) from Central Asia were collected by the authors with appropriate informed consent.12 An additional 4860 samples form part of sets published previously13, 14, 15, 16, 17, 18, 19, 20, 21 with the exclusion of population samples containing fewer than 15 individuals. A total of 5321 males from 127 populations were analysed (Supplementary Table 1 and Figure 1). In the 461 Central Asian samples up to 31 binary markers were typed hierarchically by using the SNaPshot protocol on an ABI3100 capillary electrophoresis apparatus (Applied Biosystems Inc., Foster City, CA, USA). Primers were based on ones published previously22, 23, 24 with additional primers based on published sequences.25 The binary markers define haplogroups that are represented in a maximum parsimony tree25 (Supplementary Figure 1). For comparison among data sets, some simplification of resolution was required, but the original haplogroup information was also retained and correspondence between previous nomenclatures was established.  Figure 2. Identification of unusually frequent haplotypes and associated DCs Through haplotyping of Central Asian samples (Supplementary Table 2) and a literature survey, we collected 5321 eight-locus Y-chromosomal microsatellite haplotypes belonging to 127 Asian populations (Figure 1 and Supplementary Table 1). We ranked haplotypes by frequency (Figure 2), reasoning that particularly frequent haplotypes should represent potential cores of clusters indicating past expansions of paternal lineages. The sample contained 2552 distinct haplotypes, 67% of which were unique, and 15 haplotypes (0.6%) were each present >20 times (and up to 71 times), indicating probable examples of successful transmissions (Figure 2). We focused on these 15 haplotypes and studied their spatial distributions and ages to illuminate the history of high reproductive success of their respective common ancestors. Frequent haplotypes are expected to be accompanied by neighbouring haplotypes within the same Y-SNP haplogroup that arise from them via mutation, thereby forming DCs.6 These DCs were uniformly and objectively extracted from the database using ‘Cluster Generator’. When applied to the 15 most frequent haplotypes, this led to some common haplotypes merging into clusters centred on other common haplotypes, and thus to a total of 11 DCs, whose characteristics are summarised in Table 1.  Figure 3. Characteristics of DCs A total of 2000 (37.8%) males carry Y-chromosomes belonging to these DCs, and they are remarkably widespread, with only three of the 127 populations analysed lacking any DC chromosomes; DC2 is also detected in one of the two ancient DNA population samples in our database (Krasnoyarsk_Kurgan17). Overall DC proportions in each population vary greatly from 5 to 96.7% (Supplementary Table 3). As DC4 is restricted to Korea,18 it was not considered for interpopulation analysis. For the remaining 10 DCs, we explored three characteristics for each: its geographical frequency distribution, the geographical distribution of its mean microsatellite variance to indicate its likely expansion source and its TMRCA to suggest the age of expansion. Frequency and variance are represented on maps (Figure 3 and Supplementary Figure 2), and are summarised in Table 2 and Figure 4, which also gives TMRCA estimates. Note that the dating method used, BATWING, provides estimates with large confidence intervals.  Figure 4. Our results show heterogeneity in DC features for all variables analysed: (i) Geographical pattern: two kinds of pattern are observed: six DCs display high frequencies spread over large geographic areas (DCs 1, 2, 6, 8, 10 and 14), and four DCs are more restricted to specific areas (DCs 3, 5, 8 and 12). (ii) Origin: three main source locations emerge: Silk Road/East Mongolia (DCs 1, 8 and 10), Middle East/Central Asia (DCs 2, 3 and 5) and East India/South East Asia (DCs 2, 6, 11, 12 and 14). The potential ‘source’ populations are given in Table 2. (iii) Age: mean TMRCA estimates range from 900 to 4100 YA. In considering these results, we focus on two periods: the ‘historical’ period (700–1060 CE) and the protohistorical period (2100–300 BCE), corresponding, respectively, to pre-Imperial ‘Ancient’ China,37 and the Bronze Age/Iron Age in Levantine/Inner Asia. A favourable post-Bronze Age socio-political context for successful Y-lineage transmissions We have detected 11 highly represented Y-chromosome lineages among the 5321 males analysed; altogether the DCs derived from 11 founding chromosomes represent a large proportion (38%) of the Y-chromosomes analysed, attesting to the importance of efficient Y-lineage transmission in the history of the continent. The presence of DCs with TMRCAs from 2100 BCE to 1100 CE suggests that the socio-cultural context, from the Bronze Age up to more recent historical periods, has been sufficiently favourable to ensure a continuous transmission of these different Y-lineages. Indeed, several admixture events have deeply affected the Asian gene pool during this period, involving migrant groups coming from North East and South East Asia.39 It has been argued40 that the development and impact of complex polities beginning with the Bronze Age ~2500–100 BCE in Central Asia were responsible for drastic changes in population structure, movements and organisation. These polities and early states emerged from local traditions of mobility and multiresource pastoralism, including agriculture and distributed hierarchy and administration,41, 42 with a royal or imperial form of leadership combining elements of kinship and political office.42 Archaeological evidence from the late Bronze and early Iron Ages (1400–400 BCE) confirms the existence of control hierarchies associated with wealth differences and socio-political power, even before the emergence of political entities.42 The development of these polities was accompanied by the emergence of stratified societies, recognised elite lineages and differences in power and status between individuals. High reproductive success is often associated with high social status, ‘prestigious’ men having higher intramarital fertility, lower offspring mortality9 and access to a greater than average number of wives.43 This suggests that the link between status and fitness detected in modern populations (eg, Nettle and Pollet,44 Snyder et al45 and Heyer et al46) also existed in ancient populations. For the TMRCA period from 2100 BCE to 1100 CE, the DCs are equally distributed between sedentary agriculturalist (36.8%) and pastoral nomadic populations (36.7%), indicating that uninterrupted transmission of Y-lineages over many generations could occur in both groups. Cultural transmission of fertility, reproductive success and prestige, seen in contemporaneous populations,47 could also explain the persistence of these frequent Y-lineages over time. The high variance of DC proportions among populations indicates that the ‘memes’ responsible for cultural transmission can vary greatly among populations, even when populations share similar cultural characteristics. European Journal of Human Genetics (2015) 23, 1413–1422; doi:10.1038/ejhg.2014.285; published online 14 January 2015 |
|
|
|
Post by Admin on Sept 23, 2016 21:06:39 GMT
 Figure 1 In surveys of DNA variation in Asia, we typed 2,123 men with ⩾32 markers to produce a Y haplotype for each man; these included 1,126 individuals described elsewhere (Qamar et al. 2002; Zerjal et al. 2002). Over 90% of the haplotypes showed the usual pattern (Mohyuddin et al. 2001): most males had a unique code; and the few haplotypes present in more than one individual were generally found within the same population. However, we also saw one pattern that was novel in two respects. First, there was a high frequency of a cluster of closely related lineages, collectively called the “star cluster” (fig. 1, shaded area). Second, star-cluster chromosomes were found in 16 populations throughout a large geographical area extending from Central Asia to the Pacific (fig. 2); thus, they do not result from an event specific to any single population. We can deduce the most likely time to the most recent common ancestor (TMRCA) and place of origin of this unusual lineage from the observed genetic variation. To do this, it is first necessary to distinguish star-cluster chromosomes from the remainder. For this, we used the criterion that haplotypes linked to the central one in the shaded area of the network without gaps would be included (fig. 1). We then used two approaches to calculate a TMRCA for the star-cluster chromosomes. The program BATWING (Wilson and Balding 1998) uses models of both mutation and population processes, which were specified as described elsewhere (Qamar et al. 2002). With this program, we estimated ∼1,000 years for the TMRCA (95% confidence interval limits ∼700–1,300 years). The use of alternative demographic models with constant or exponentially increasing population size changed the estimate by <10%. A method that does not consider population structure (Morral et al. 1994), ρ, suggested ∼860 (∼590–1,300) years. In both calculations, we assumed a generation time of 30 years. The origin was most likely in Mongolia, where the largest number of different star-cluster haplotypes is found (fig. 1). Thus, a single male line, probably originating in Mongolia, has spread in the last ∼1,000 years to represent ∼8% of the males in a region stretching from northeast China to Uzbekistan. If this spread were due to a general population expansion, we would expect to find multiple lineages with the same characteristics of high frequency and presence in multiple populations, but we do not (Zerjal et al. 2002). The star-cluster pattern is unique. This rise in frequency, if spread evenly over ∼34 generations, would require an average increase by a factor of ∼1.36 per generation and is thus comparable to the most extreme selective events observed in natural populations, such as the spread of melanic moths in 19th-century England in response to industrial pollution (Edleston 1865). We evaluated whether it could have occurred by chance. If the population growth rate is known, it is possible to test whether the observed frequency of a lineage is consistent with its level of variation, assuming neutrality (Slatkin and Bertorelle 2001). Using this method, we estimated the chance of finding the low degree of variation observed in the star cluster, with a current frequency of ∼8%, under neutral conditions. Even with the demographic model most likely to lead to rapid increase of the lineage, double exponential growth, the probability was <10−237; if the mutation rate were 10 times lower, the probability would still be <10−10. Thus, chance can be excluded: selection must have acted on this haplotype.  Figure 2 Could biological selection be responsible? Although this possibility cannot be entirely ruled out, the small number of genes on the Y chromosome and their specialized functions provide few opportunities for selection (Jobling and Tyler-Smith 2000). It is therefore necessary to look for alternative explanations. Increased reproductive fitness, transmitted socially from generation to generation, of males carrying the same Y chromosome would lead to the increase in frequency of their Y lineage, and this effect would be enhanced by the elimination of unrelated males. Within the last 1,000 years in this part of the world, these conditions are met by Genghis (Chingis) Khan (c. 1162–1227) and his male relatives. He established the largest land empire in history and often slaughtered the conquered populations, and he and his close male relatives had many children. Although the Mongol empire soon disintegrated as a political unit, his male-line descendants ruled large areas of Asia for many generations. These included China, where the Yüan Dynasty emperors remained in power until 1368, after which the Mongols continued to dominate the country north of the Great Wall for several more centuries, and the region west to the Aral Sea, where the Chaghatai Khans ruled. Although their power diminished over time, they remained at Kashghar near the Kyrgyzstan/China border until the middle of the 17th century (Morgan 1986).  It is striking that the boundary of the Mongol empire when Genghis Khan died (fig. 2), which also corresponds to the boundaries of the regions controlled by later Khans, matches the distribution of star-cluster chromosomes closely, with one exception: the Hazaras. We, therefore, wished to compare Genghis Khan’s Y profile with the star cluster. It is not possible to examine his remains directly, but history provides an alternative. The Hazaras of Pakistan have a Mongol origin (Qamar et al. 2002), and many consider themselves to be direct male-line descendants of Genghis Khan. A genealogy documenting these links has been constructed from their oral history (Mousavi 1998). A large proportion of the Hazara profiles do indeed lie in the star cluster, which is not otherwise seen in Pakistan (fig. 2), thus supporting their oral tradition and suggesting that Genghis Khan carried the star-cluster haplotype.  The Y chromosome of a single individual has spread rapidly and is now found in ∼8% of the males throughout a large part of Asia. Indeed, if our sample is representative, this chromosome will be present in about 16 million men, ∼0.5% of the world's total. The available evidence suggests that it was carried by Genghis Khan. His Y chromosome would obviously have had ancestors, and our best estimate of the TMRCA of star-cluster chromosomes lies several generations before his birth. Several scenarios, which are not mutually exclusive, could explain its rapid spread: (1) all populations carrying star-cluster chromosomes could have descended from a common ancestral population in which it was present at high frequency; (2) many or most Mongols at the time of the Mongol empire could have carried these chromosomes; (3) it could have been restricted to Genghis Khan and his close male-line relatives, and this specific lineage could have spread as a result of their activities. Explanation 1 is unlikely because these populations do not share other Y haplotypes, and explanation 2 is difficult to reconcile with the high Y-haplotype diversity of modern Mongolians (Zerjal et al. 2002). The historically documented events accompanying the establishment of the Mongol empire would have contributed directly to the spread of this lineage by Genghis Khan and his relatives, but perhaps as important was the establishment of a long-lasting male dynasty. This scenario shows selection acting on a group of related men; group selection has been much discussed (Wilson and Sober 1994) and is distinguished by the property that the increased fitness of the group is not reducible to the increased fitness of the individuals. It is unclear whether this is the case here. Our findings nevertheless demonstrate a novel form of selection in human populations on the basis of social prestige. A founder effect of this magnitude will have influenced allele frequencies elsewhere in the genome: mitochondrial DNA lineages will not be affected, since males do not transmit their mitochondrial DNA, but, in the simplest models, the founder male will have been the ancestor of each autosomal sequence in ∼4% of the population and X-chromosomal sequence in ∼2.7%, with implications for the medical genetics of the region. Large-scale changes to patterns of human genetic variation can occur very quickly. Although local influences of this kind may have been common in human populations, it is, perhaps, fortunate that events of this magnitude have been rare. AJHG Volume 72, Issue 3, p717–721, March 2003 |
|
|
|
Post by Admin on Sept 27, 2016 21:00:15 GMT
 Figure 1 Frequency distribution of Y-chromosomal haplotypes in East Asia. The two most frequent haplotypes identified by the 15 loci Previously, 1,003 males representing 28 populations from China, Mongolia, Korea, and Japan were typed with 16 Y STRs (Zerjal et al. 2003). The frequency distribution of 15-element haplotypes, constructed without DYS19 since duplications of this locus are common in this data set, shows that most haplotypes (621 [84%] of 736) are present in a single individual and that haplotypes shared by two or more individuals become progressively rarer, so that no haplotype is shared by nine individuals (fig. 1). Five haplotypes, however, fell outside this distribution pattern and were unexpectedly frequent in East Asian populations, having been found in 11, 13, 15, 23, and 27 individuals. The first three were found predominantly in a single population or two nearby populations and, thus, could have been spread by local drift in populations of small size. The other two, however, were more widespread. The haplotype present in 23 individuals was found in eight populations (and in more that are outside the area considered here) and is the “star cluster” described elsewhere (Zerjal et al. 2003). The most frequent haplotype in this sample, designated the “Manchu haplotype” for reasons described below, was present in 27 individuals belonging to seven populations. These two high-frequency haplotypes differ by only four steps, which raises the question of whether their prevalence might have a common cause. We show below, however, that they belong to different haplogroups and, thus, that their recent expansions must be independent. Here, we examine the distribution and likely origin of the second unusual haplotype. The 1,003 males were tested with 45 binary markers representing the known Y-chromosomal SNP variation in this part of the world (Zerjal et al. 2003; results not shown), and all Manchu haplotype chromosomes were found to carry the derived allele of M48 and, thus, fall into haplogroup C3c. A median-joining network (Bandelt et al. 1999) of this haplogroup showed that closely related chromosomes that might share a common origin were also present, forming a “Manchu cluster” (fig. 2a; data in a tab-delimited ASCII file [online only] that can be imported into a spreadsheet). However, it was not obvious where the boundaries of this cluster lay—chromosomes zero, one, two, three, four, and five steps away from the center were present 27, 7, 3, 5, 3, and 3 times, respectively, showing a decrease in frequency but not a discontinuity. We wished to define the cluster so that we could then map its geographical distribution and estimate its time to the most recent common ancestor (TMRCA), so we needed a definition independent of geography and assumed diversity. We therefore typed the C3c chromosomes with 46 new simple Y STRs (Kayser et al. 2004). The resulting 61-STR network shows, as was expected because of the larger number of markers, considerable resolution within the Manchu cluster and population-specific substructure (fig%. 2b). We defined the Manchu cluster as the set of chromosomes linked to the modal haplotype (arrow in fig%. 2b), allowing a run of up to three empty nodes. According to this definition, the cluster contains 33 chromosomes: 27/27 at the center of the 15-element network, 5/7 one step away, 1/3 two steps away, and none of the more distant chromosomes (fig%. 2a). With this definition, we could examine the distribution and time depth of the cluster.  Figure 2 Median-joining network of haplogroup C3c Y chromosomes from East Asia. a, Network constructed using 15 Y STRs (Network 4.1.0.9; ASCII file [online only]). Manchu cluster chromosomes were present in seven populations: Xibe, Outer Mongolians, Inner Mongolians, Ewenki, Oroqen, Manchu, and Hezhe (fig. 3). With the exception of the Xibe, these populations are all located in northeastern China or Mongolia, and the Xibe migrated to their present location in western China from northeastern China in 1764 (Du and Yip 1993). These findings, therefore, suggest that the cluster originated and spread locally in northeastern China/Mongolia and that this happened before the time of the Xibe migration. A more precise estimate of TMRCA (mean ± SD) of the Manchu cluster, from the program Network 4.1.0.9 (Bandelt et al. 1999), was 590 ± 340 years or 220 ± 130 years, depending on whether the inferred (Zhivotovsky et al. 2004) or observed (Kayser et al. 2000; Dupuy et al. 2004) mutation rates were used. The former rate is calibrated for time periods of ∼1,000 years, and the latter is a direct measurement over single-generation times, so the most likely TMRCA is intermediate between these values and will be referred to as ∼500 years. We then wished to know whether the haplotype sharing between populations and the rapid expansion were characteristics of the populations as a whole, resulting from the general demography of the area, or were specific to the Manchu cluster. The proportion of 15-STR haplotypes (excluding the Manchu cluster) shared between populations and the ρ distance (the number of steps between a 15-element haplotype in one population and the closest haplotype in a second population, averaged over all haplotypes [Helgason et al. 2000]) were therefore calculated for Mongolian and northeastern Chinese populations (table 1). These measures were not significantly different between populations that carried the Manchu cluster and those that lacked it, showing that the Manchu cluster is exceptional, in that its sharing between populations is a lineage-specific phenomenon rather than a general feature of all lineages in these populations.  Figure 3 Geographical distribution of Manchu cluster chromosomes. Circles, Populations sampled, with area proportional to sample size. Filled sectors, Manchu clusters. We reasoned that the events leading to the spread of this lineage might have been recorded in the historical record, as well as in the genetic record. The spread must have occurred after the cluster's TMRCA (∼500 years ago, corresponding to about a.d. 1500) and, most likely, before the Xibe migration in 1764. Notable features are the occurrence of the lineage in seven different populations but its apparent absence from the most populous Chinese ethnic group, the Han. A major historical event took place in this part of the world during this period—namely, the Manchu conquest of China and the establishment of the Qing dynasty, which ruled China from 1644 to 1912. This dynasty was founded by Nurhaci (1559–1626) and was dominated by the Qing imperial nobility, a hereditary class consisting of male-line descendants of Nurhaci's paternal grandfather, Giocangga (died 1582), with >80,000 official members by the end of the dynasty (Elliott 2001). The nobility were highly privileged; for example, a ninth-rank noble annually received ∼11 kg of silver and 22,000 liters of rice and maintained many concubines. A central part of the Qing social system was the army, the Eight Banners, which was made up of separate Manchu, Mongolian, and Chinese (Han) Eight Banners. The nobility occupied high ranks in the Manchu Eight Banners but not in the Mongolian or Chinese Eight Banners; the Manchu Eight Banners were recruited from the Manchu, Mongolian, Daur, Oroqen, Ewenki, Xibe, and a few other populations. A social mechanism was thus established that would have led to the increase of the specific Y lineage carried by Giocangga and Nurhaci and to its spread into a limited number of populations. We suggest that this lineage was the Manchu lineage. Our hypothesis could be tested by examining the descendants of the Qing nobility (Li 1997). Unfortunately, extensive warfare during the 20th century and the Cultural Revolution (1966–1976) led to enormous social upheaval, during which descent from the nobility was usually hidden and relevant documents were destroyed. As a result, very few well-attested descendants are known, and they were not available for testing. Thus, our hypothetical explanation remains unproven. Nevertheless, it has strong circumstantial support. The 80,000 nobility at the end of the Qing Dynasty would have represented ∼0.8% of the non-Han population of China at that time and, at this frequency, should contribute ∼5 chromosomes to our sample. The number of males carrying this chromosome would have greatly exceeded the officially recognized number of nobility, so the number 5 should be multiplied several times, and the nobility's chromosomes should be distributed among the Manchu Eight Banner populations. The lineage should, therefore, be detectable in our data set as a widespread, high-frequency northern haplotype. The Manchu cluster is the only lineage that meets these requirements. AJHG Volume 77, Issue 6, p1112–1116, December 2005 |
|

