Post by Admin on Jun 29, 2020 20:14:30 GMT
To gain further insights into the pathogens associated with this historical genitourinary infection, we pooled reads from all nodule DNA libraries, mapped and reconstructed the ancient S. saprophyticus and G. vaginalis genomes and analyzed them in conjunction with existing and newly acquired genomic data from extant and historical organisms (Supplementary file 1H,I).
We used a combination of paired-end reference guided assembly and iterative assembly to reconstruct a nearly complete genome of S. saprophyticus Troy, including >100 Kb of novel sequence compared to reference strain ATCC 15305. The genome is 2,471,881 bp long, with an average unique coverage depth of 298.6x (Figure 4—figure supplement 3), which represents an unprecedented, detailed and complete picture of an ancient pathogen genome from shotgun sequencing data. We also reconstructed a 22.6 Kb plasmid, pSST1.
We were unable to reconstruct a contiguous G. vaginalis genome due to high variability in coverage and lack of synteny in both ancient and modern genomic data (Ahmed et al., 2012). Instead, we used a de novo approach to reconstruct G. vaginalis Troy gene content using reads that mapped to the annotated coding regions of all available G. vaginalis genomes. This enabled us to assess the gene content of our ancient genome compared to the modern strains. Using this method, we recovered 1187 unique contigs (total length 1,435,761 bp) corresponding to 972 annotated genes and an average unique coverage depth of 57.0x (Figure 3—figure supplement 3).
Our sample of 35 isolates of G. vaginalis was grouped into four previously defined clades (Figure 3, Figure 3—figure supplement 4), which have been proposed to represent distinct species (Ahmed et al., 2012). G. vaginalis Troy sits within Clade 1, among vaginal and endometrial isolates collected from both healthy women and patients with bacterial vaginosis. Interestingly, the 800-year-old sample from Troy (Turkey) falls within contemporary genetic diversity (Supplementary file 1I).
Figure 3
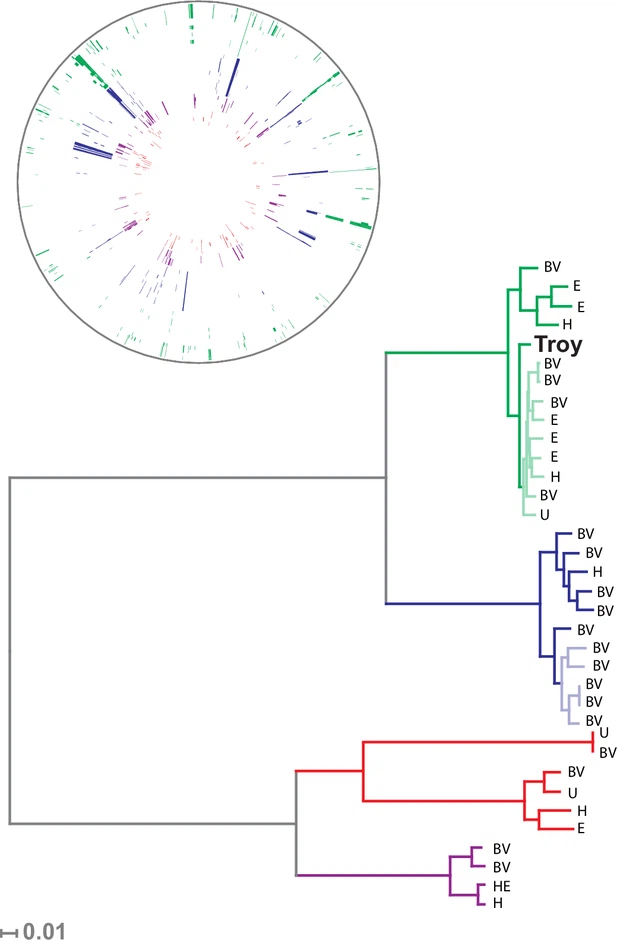
Consistent with prior reports (Ahmed et al., 2012), we identified extensive impacts of lateral gene transfer (LGT) on G. vaginalis diversity (Figure 3). Even in the core genome alignment, which contains just 44% of per-isolate gene content, we estimate that 20% of sites have been affected by recombination. This high rate of recombination may help to explain the remarkable preservation of genetic diversity in G. vaginalis. A recent study of Helicobacter pylori, which has similarly high rates of LGT, found that genetic diversity within the species has been preserved for more than five thousand years (Maixner et al., 2016).
We discovered two distinct clades of S. saprophyticus (Figure 4, Figure 4—figure supplement 4), one of which (Clade P) appears to be more strongly associated with pathogenicity and includes our ancient S. saprophyticus Troy. Nineteen of twenty veterinary and human clinical isolates belong to Clade P, an association that was statistically significant (Appendix). A second clade (Clade E) is made up of food and environmental isolates of S. saprophyticus, as well as a human UTI isolate from Japan.
Figure 4
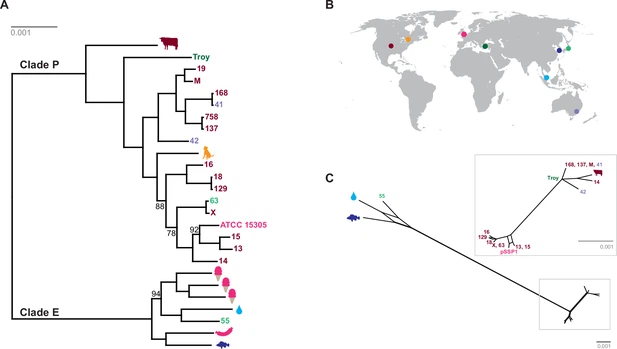
Plasmids similar to S. saprophyticus Troy pSST1 were present in isolates from both clades. The relationships among plasmid sequences from S. saprophyticus Troy and other members of Clade P were distinct from those of the core genome; we also found evidence of recombination among pSST1-like plasmids (Figure 4C, Appendix).
A long branch separates Clade P pSST1 from those of Clade E (Figure 4C), recapitulating their relationship on the core genome phylogeny. This was also true of pSSP2, the only other plasmid we identified in both Clades P and E (but not S. saprophyticus Troy; Figure 4—figure supplement 5). These observations suggest plasmids are more readily exchanged within Clades P and E than between them. This could indicate that Clades P and E are spatially segregated, that there are mechanistic barriers to plasmid exchange between clades, or that epistatic interactions reinforce clade separation of these mobile elements.
The human clinical isolates in Clades P and E are nested within the phylogeny with more divergent lineages associated with other animals. This suggests that the most recent common ancestor (MRCA) of S. saprophyticus may not have been human-associated. This is in stark contrast to the major pathogen in the genus, Staphylococcus aureus, where phylogenetic studies suggest that the MRCA of human and livestock-associated lineages had a human host (Fitzgerald, 2012; Weinert et al., 2012; Shepheard et al., 2013). S. aureus is strongly associated with its niche on the human body and is transmitted primarily from person-to-person. S. saprophyticus, by contrast, appears to be a generalist that colonizes a range of environments.
Several lines of evidence also indicate that humans acquire S. saprophyticus infection from the environment. In northern climates, there is marked seasonal variation in the incidence of S. saprophyticus UTI (Rupp et al., 1992; Hovelius and Mårdh, 1984; Ringertz and Torssander, 1986; Hedman et al., 1993; Widerström et al., 2007), whereas there is no evidence of seasonality in Mediterranean climates (Schneider and Riley, 1996). S. saprophyticus can be identified in environmental samples, with a strong seasonal peak that occurs just before peak rates of S. saprophyticus UTI in northern climates (Hedman et al., 1993; Soge et al., 2009). Molecular epidemiological surveys also suggest S. saprophyticus is primarily acquired from an environmental reservoir, rather than as a result of person-to-person transmission (Widerström et al., 2007; Widerström et al., 2012). These observations suggest that the bacteria cycle between host-associated and environmental stages, with seasonal climatic effects on their abundance in the environment.
The length of the branch leading to S. saprophyticus Troy is similar to those leading to the other tips (Figure 4A), suggesting there is little temporal signal in the phylogeny. Calibrated phylogenetic analyses (Appendix) confirmed the absence of temporal signal, which precludes reliable estimation of the rate of substitution or divergence times for S. saprophyticus.
A mixed environmental, commensal and pathogenic niche may in part explain the absence of temporal structure in our sample of S. saprophyticus. Selection pressures and generation times are likely to differ between free-living and host-associated stages, which can obscure temporal signals in genetic data (Bromham, 2009). In addition to producing rate variability, periods of dormancy in the environment – e.g. during the winter in northern climates, as suggested by seasonal patterns in cultivability – would be predicted to lower the overall rate at which S. saprophyticus evolves (Bromham, 2009; Weinert et al., 2015). The 800 year interval between S. saprophyticus Troy and the other tips may simply be too short relative to the overall depth of the tree to allow reliable rate inference.
Notably, all human-associated isolates of S. saprophyticus in Clade P form a monophyletic group, to which the bovine mastitis strain falls basally; there are no modern human pathogenic representatives of the S. saprophyticus Troy lineage. This may mean that the ecology of S. saprophyticus differed in the Byzantine world, with human infections arising from a different reservoir of bacteria than they do today. S. saprophyticus is readily cultured from the environment around livestock (Hedman et al., 1993; Cherif-Antar et al., 2016), and Byzantine era peasants in Anatolia typically shared their households with cattle (Lefort, 2007). This and other historical settings are likely to have facilitated spillover events and, perhaps, the circulation of bacteria that were adapted to both livestock and humans.
Based on the available data, it is not possible to determine whether the human clinical isolate nested among environmental and food-associated bacteria in Clade E represents a spillover or a second emergence into humans. In either event, it appears that S. saprophyticus can transition to a human pathogenic niche with relative ease. We did not identify any gene content uniquely shared (or absent) among the pathogenic strains in our sample, which suggests that pleiotropy underlies S. saprophyticus’ flexible association with diverse niches. For many bacterial genera, genetic distances between free-living organisms and pathogens are larger than observed here, and pathogen emergence is a singular event characterized by genomic decay and loss of functions required outside the pathogenic niche (Parkhill et al., 2001; Larsson et al., 2009; Reuter et al., 2014). More studies and wider sampling are needed to fully characterize the niche of S. saprophyticus, but our observations reinforce the notion that the adaptive paths to bacterial virulence are more diverse than has previously been appreciated.
Complications of pregnancy and childbirth are major causes of morbidity and mortality worldwide and new threats to maternal health continue to emerge (WHO et al., 2014; Mlakar et al., 2016). Our analyses of the remains of a woman who died in Late Byzantine Troy connect her to this broad historical and epidemiological phenomenon. Her infection was associated with exuberant calcification of the placenta, which replicated maternal, fetal, and bacterial cells in calcium phosphate minerals and preserved a high resolution molecular portrait of their contents. S. saprophyticus, an organism the decedent is likely to have acquired from her environment, and G. vaginalis, a member of the native human biota, are the dominant bacterial species of the infection. S. saprophyticus Troy belongs to a lineage that appears to be uncommonly associated with human disease in the modern world, whereas G. vaginalis Troy nests among modern commensal and pathogenic strains on its phylogeny. This highlights the complexity of virulence as a bacterial trait and a potential role of interactions among bacterial species in shaping pathologic outcomes of infection.
We used a combination of paired-end reference guided assembly and iterative assembly to reconstruct a nearly complete genome of S. saprophyticus Troy, including >100 Kb of novel sequence compared to reference strain ATCC 15305. The genome is 2,471,881 bp long, with an average unique coverage depth of 298.6x (Figure 4—figure supplement 3), which represents an unprecedented, detailed and complete picture of an ancient pathogen genome from shotgun sequencing data. We also reconstructed a 22.6 Kb plasmid, pSST1.
We were unable to reconstruct a contiguous G. vaginalis genome due to high variability in coverage and lack of synteny in both ancient and modern genomic data (Ahmed et al., 2012). Instead, we used a de novo approach to reconstruct G. vaginalis Troy gene content using reads that mapped to the annotated coding regions of all available G. vaginalis genomes. This enabled us to assess the gene content of our ancient genome compared to the modern strains. Using this method, we recovered 1187 unique contigs (total length 1,435,761 bp) corresponding to 972 annotated genes and an average unique coverage depth of 57.0x (Figure 3—figure supplement 3).
Our sample of 35 isolates of G. vaginalis was grouped into four previously defined clades (Figure 3, Figure 3—figure supplement 4), which have been proposed to represent distinct species (Ahmed et al., 2012). G. vaginalis Troy sits within Clade 1, among vaginal and endometrial isolates collected from both healthy women and patients with bacterial vaginosis. Interestingly, the 800-year-old sample from Troy (Turkey) falls within contemporary genetic diversity (Supplementary file 1I).
Figure 3
Consistent with prior reports (Ahmed et al., 2012), we identified extensive impacts of lateral gene transfer (LGT) on G. vaginalis diversity (Figure 3). Even in the core genome alignment, which contains just 44% of per-isolate gene content, we estimate that 20% of sites have been affected by recombination. This high rate of recombination may help to explain the remarkable preservation of genetic diversity in G. vaginalis. A recent study of Helicobacter pylori, which has similarly high rates of LGT, found that genetic diversity within the species has been preserved for more than five thousand years (Maixner et al., 2016).
We discovered two distinct clades of S. saprophyticus (Figure 4, Figure 4—figure supplement 4), one of which (Clade P) appears to be more strongly associated with pathogenicity and includes our ancient S. saprophyticus Troy. Nineteen of twenty veterinary and human clinical isolates belong to Clade P, an association that was statistically significant (Appendix). A second clade (Clade E) is made up of food and environmental isolates of S. saprophyticus, as well as a human UTI isolate from Japan.
Figure 4
Plasmids similar to S. saprophyticus Troy pSST1 were present in isolates from both clades. The relationships among plasmid sequences from S. saprophyticus Troy and other members of Clade P were distinct from those of the core genome; we also found evidence of recombination among pSST1-like plasmids (Figure 4C, Appendix).
A long branch separates Clade P pSST1 from those of Clade E (Figure 4C), recapitulating their relationship on the core genome phylogeny. This was also true of pSSP2, the only other plasmid we identified in both Clades P and E (but not S. saprophyticus Troy; Figure 4—figure supplement 5). These observations suggest plasmids are more readily exchanged within Clades P and E than between them. This could indicate that Clades P and E are spatially segregated, that there are mechanistic barriers to plasmid exchange between clades, or that epistatic interactions reinforce clade separation of these mobile elements.
The human clinical isolates in Clades P and E are nested within the phylogeny with more divergent lineages associated with other animals. This suggests that the most recent common ancestor (MRCA) of S. saprophyticus may not have been human-associated. This is in stark contrast to the major pathogen in the genus, Staphylococcus aureus, where phylogenetic studies suggest that the MRCA of human and livestock-associated lineages had a human host (Fitzgerald, 2012; Weinert et al., 2012; Shepheard et al., 2013). S. aureus is strongly associated with its niche on the human body and is transmitted primarily from person-to-person. S. saprophyticus, by contrast, appears to be a generalist that colonizes a range of environments.
Several lines of evidence also indicate that humans acquire S. saprophyticus infection from the environment. In northern climates, there is marked seasonal variation in the incidence of S. saprophyticus UTI (Rupp et al., 1992; Hovelius and Mårdh, 1984; Ringertz and Torssander, 1986; Hedman et al., 1993; Widerström et al., 2007), whereas there is no evidence of seasonality in Mediterranean climates (Schneider and Riley, 1996). S. saprophyticus can be identified in environmental samples, with a strong seasonal peak that occurs just before peak rates of S. saprophyticus UTI in northern climates (Hedman et al., 1993; Soge et al., 2009). Molecular epidemiological surveys also suggest S. saprophyticus is primarily acquired from an environmental reservoir, rather than as a result of person-to-person transmission (Widerström et al., 2007; Widerström et al., 2012). These observations suggest that the bacteria cycle between host-associated and environmental stages, with seasonal climatic effects on their abundance in the environment.
The length of the branch leading to S. saprophyticus Troy is similar to those leading to the other tips (Figure 4A), suggesting there is little temporal signal in the phylogeny. Calibrated phylogenetic analyses (Appendix) confirmed the absence of temporal signal, which precludes reliable estimation of the rate of substitution or divergence times for S. saprophyticus.
A mixed environmental, commensal and pathogenic niche may in part explain the absence of temporal structure in our sample of S. saprophyticus. Selection pressures and generation times are likely to differ between free-living and host-associated stages, which can obscure temporal signals in genetic data (Bromham, 2009). In addition to producing rate variability, periods of dormancy in the environment – e.g. during the winter in northern climates, as suggested by seasonal patterns in cultivability – would be predicted to lower the overall rate at which S. saprophyticus evolves (Bromham, 2009; Weinert et al., 2015). The 800 year interval between S. saprophyticus Troy and the other tips may simply be too short relative to the overall depth of the tree to allow reliable rate inference.
Notably, all human-associated isolates of S. saprophyticus in Clade P form a monophyletic group, to which the bovine mastitis strain falls basally; there are no modern human pathogenic representatives of the S. saprophyticus Troy lineage. This may mean that the ecology of S. saprophyticus differed in the Byzantine world, with human infections arising from a different reservoir of bacteria than they do today. S. saprophyticus is readily cultured from the environment around livestock (Hedman et al., 1993; Cherif-Antar et al., 2016), and Byzantine era peasants in Anatolia typically shared their households with cattle (Lefort, 2007). This and other historical settings are likely to have facilitated spillover events and, perhaps, the circulation of bacteria that were adapted to both livestock and humans.
Based on the available data, it is not possible to determine whether the human clinical isolate nested among environmental and food-associated bacteria in Clade E represents a spillover or a second emergence into humans. In either event, it appears that S. saprophyticus can transition to a human pathogenic niche with relative ease. We did not identify any gene content uniquely shared (or absent) among the pathogenic strains in our sample, which suggests that pleiotropy underlies S. saprophyticus’ flexible association with diverse niches. For many bacterial genera, genetic distances between free-living organisms and pathogens are larger than observed here, and pathogen emergence is a singular event characterized by genomic decay and loss of functions required outside the pathogenic niche (Parkhill et al., 2001; Larsson et al., 2009; Reuter et al., 2014). More studies and wider sampling are needed to fully characterize the niche of S. saprophyticus, but our observations reinforce the notion that the adaptive paths to bacterial virulence are more diverse than has previously been appreciated.
Complications of pregnancy and childbirth are major causes of morbidity and mortality worldwide and new threats to maternal health continue to emerge (WHO et al., 2014; Mlakar et al., 2016). Our analyses of the remains of a woman who died in Late Byzantine Troy connect her to this broad historical and epidemiological phenomenon. Her infection was associated with exuberant calcification of the placenta, which replicated maternal, fetal, and bacterial cells in calcium phosphate minerals and preserved a high resolution molecular portrait of their contents. S. saprophyticus, an organism the decedent is likely to have acquired from her environment, and G. vaginalis, a member of the native human biota, are the dominant bacterial species of the infection. S. saprophyticus Troy belongs to a lineage that appears to be uncommonly associated with human disease in the modern world, whereas G. vaginalis Troy nests among modern commensal and pathogenic strains on its phylogeny. This highlights the complexity of virulence as a bacterial trait and a potential role of interactions among bacterial species in shaping pathologic outcomes of infection.