|
|
Post by Admin on Oct 22, 2013 21:08:34 GMT
 A record of Neanderthal history, thought to be lost a century ago, has been re-discovered in Jersey. The island is believed to be one of the last places the Neanderthals lived after teeth were discovered on its Cotte de St Brelade cave (pictured) in 1910 Modern humans were living in Italy and the UK as far back as 41,000-45,000 years ago, putting them in contact with European Neanderthals who persisted on the continent for many millennia after these dates. On the Rock of Gibraltar, Neanderthals could possibly have survived around until as recently as 28,000 years ago before finally dying out. Neanderthals also lived on the Channel Island of Jersey from around 250,000 years ago until between 100,000 and 47,000 years ago and the sequence of sediments at the site of La Cotte de St Brelade dates between 100,000 and 47,000 years old, indicating that Neanderthal teeth which were discovered at the site in 1910 were younger than previously thought. In 2011, a programme of field research was undertaken to effect the stabilization of an unstable section in the West Ravine at the key Neanderthal occupation site of La Cotte de St Brelade on the Channel Island of Jersey. As part of this essential remedial work the threatened section was analysed to characterize its archaeological and palaeoenvironmental potential as well provide optically stimulated luminescence (OSL) dates. The work determined, through two concordant OSL dating programmes, that the section formed part of an extensive sequence of sedimentation spanning >105 to <48 ka. Furthermore, reanalysis of the archive determined that the sediment sequence examined contained the stratigraphic equivalent of deposits lying below those that have previously produced Neanderthal fossils. Through our work, we can now constrain these younger sediments to being younger than 48 ka. The combined results suggest that this sequence now represents the recovery of an extensive dataset, thought lost to science through complete excavation, which holds the potential to throw light on the disappearance of Neanderthal populations from the Atlantic-edge outpost on the north-west frontier of their world. onlinelibrary.wiley.com/doi/10.1002/jqs.2669/abstractThe evidence for Neanderthal lithic technology is reviewed and summarized for four caves on The Rock of Gibraltar: Vanguard, Beefsteak, Ibex and Gorham’s. Some of the observed patterns in technology are statistically tested including raw material selection, platform preparation, and the use of formal and expedient technological schemas. The main parameters of technological variation are examined through detailed analysis of the Gibraltar cores and comparison with samples from the classic Mousterian sites of Le Moustier and Tabun C. The Gibraltar Mousterian, including the youngest assemblage from Layer IV of Gorham’s Cave, spans the typical Middle Palaeolithic range of variation from radial Levallois to unidirectional and multi-platform flaking schemas, with characteristic emphasis on the former. A diachronic pattern of change in the Gorham’s Cave sequence is documented, with the younger assemblages utilising more localized raw material and less formal flaking procedures. We attribute this change to a reduction in residential mobility as the climate deteriorated during Marine Isotope Stage 3 and the Neanderthal population contracted into a refugium. 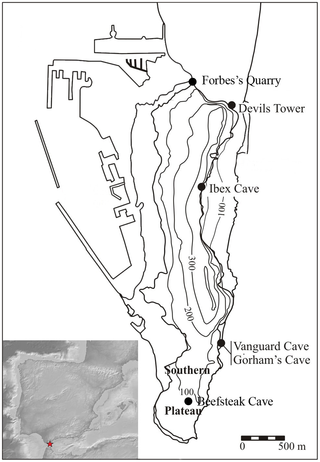 Figure 1. The location of sites mentioned in the text. Figure 1. The location of sites mentioned in the text.
Inset: the location of Gibraltar on the Iberian Peninsula.The Rock of Gibraltar is a limestone klippe peninsula at the southern tip of Iberia (Figure 1) and represents the south-western extremity of the Neanderthal range. Both wave and solutional erosion have created a series of caves in the klippe, particularly on its more exposed eastern side, which were inhabited by Neanderthals and then Homo sapiens over the last 100 thousand years. Gibraltar is home to some of the world’s most significant Neanderthal sites. The region is historically significant as one of the first discoveries of Neanderthal skeletal remains was made in Forbes Quarry in 1848 [1], [2]. Important dietary information has been obtained from the Gibraltar caves, including the exploitation of a range of terrestrial and marine species unparalleled at other Neanderthals sites [3], [4]. Gibraltar also boasts having the youngest Mousterian sites in Europe, suggesting that the area served as a refugium for the final Neanderthals [5], [6]. Bio-climatic modelling indicates that the favoured habitats of the southern Iberian Neanderthals became fragmented during MIS3 separating coastal and upland populations [56], with much of the interior of Iberia becoming arid [67]. The MIS3 occupation of Gorham’s Cave may represent a Neanderthal population which foraged locally along the coast and did not exploit inland resources to the same extent as their predecessors had done. Indeed, the low seas level stands of MIS3 would have opened up new shore habitats immediately in front of Gibraltar [58], [59]. This may explain why the technology becomes increasingly expedient and made on more local materials during the later occupation phases at Gorham’s, reflecting reduced residential mobility and greater emphasis on foraging on and around The Rock. Waechter’s layer G has a far higher artefact density than the underlying layers, suggesting more intensive occupation, perhaps as a result of a reduced residential mobility pattern [68]. An increase in the quantity of charred material in the MIS3 occupation of Gorham’s also indicates more intensive hominin occupation at this time [69]. Parallels may be found with MIS3 Neanderthals populations elsewhere. In the southern Caucasus the environment was stable and diverse, like Gibraltar, and also did not suffer the MIS3 deterioration to the same extent as surrounding regions [70]. In the late Middle Palaeolithic of the southern Caucasus, prior to replacement by Homo sapiens c. 37kya, there was also a reduction in Neanderthal range size with far fewer exotic materials being exploited than in the earlier Middle Palaeolithic [20]. In the Middle Palaeolithic of Latium, Italy, the onset of MIS3 coincided with a reduction in the import of exotic materials and a shift away from radially prepared cores for striking larger flakes, to bidirectional small flake cores [19]. Gibraltar has been hypothesized to be one of the last refuges of the Neanderthals with a date of 28 kya for the youngest Mousterian occupation in Layer IV of Gorham’s Cave [5] [6]. This young age has been challenged partly on technological grounds with the suggestion that the Layer IV occupation of Gorham’s actually represents the early Upper Palaeolithic rather than the latest Mousterian [35]. However, there are no blade cores, or even blade scars on the cores from Layer IV. There is also continuity between Layer IV and the older members of Gorham’s Cave in the use of large rounded quartzite cobbles as single platform cores and in the production of small discoidal cores on chert. In accordance with previous analyses [12] we must therefore assign the Layer IV artefacts to the Mousterian. The absence of any Levallois cores from this final occupation could reflect a dwindling population in which the expertise required for this most complex of Middle Palaeolithic technologies has been lost (sensu Henrich, [71]), but larger samples are needed to test this hypothesis. The Mousterian record from the Gibraltar caves provides a rich sequence of Neanderthal occupation in an optimal habitat. The high biodiversity and stability of the Gibraltar climate may have allowed this region to act as a refugium for the last surviving Neanderthals [63], [72], [73]. As the climate deteriorated during MIS3 the technological response of the Neanderthals was to use more expedient flaking strategies on locally available material, reflecting a reduction in mobility and a contraction into the core zone of the refugium. www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0065185 |
|
|
|
Post by Admin on Oct 24, 2013 22:47:33 GMT
 According to written sources, Roma (Romanies, Gypsies) arrived in the Balkans around 1,000 years ago from India and have subsequently spread through several parts of Europe. Genetic data, particularly from the Y chromosome, have supported this model, and can potentially refine it. We now provide an analysis of Y-chromosomal markers from five Roma and two non-Roma populations (N = 787) in order to investigate the genetic relatedness of the Roma population groups to one another, and to gain further understanding of their likely Indian origins, the genetic contribution of non-Roma males to the Roma populations, and the early history of their splits and migrations in Europe. The two main sources of the Roma paternal gene pool were identified as South Asian and European. The reduced diversity and expansion of H1a-M82 lineages in all Roma groups imply shared descent from a single paternal ancestor in the Indian subcontinent. The Roma paternal gene pool also contains a specific subset of E1b1b1a-M78 and J2a2-M67 lineages, implying admixture during early settlement in the Balkans and the subsequent influx into the Carpathian Basin. Additional admixture, evident in the low and moderate frequencies of typical European haplogroups I1-M253, I2a-P37.2, I2b-M223, R1b1-P25, and R1a1-M198, has occurred in a more population-specific manner. Haplogroup I (M170) is widespread over Europe but virtually absent elsewhere, including the Near East, suggesting that it arose in Europe (Rootsi et al., 2004). I1-M253 chromosomes were detected at relatively high frequencies in all six Roma groups compared, and 61.8% of these males share a modal haplotype (Fig. 1F), suggesting a common origin of the Roma as well. According to Gusma˜o et al. (2008), the I1-M253 marker occurred at different proportions in Roma groups compared to their host populations, but can be explained by genetic drift. The dispersed network pattern observed for I2a-P37.2 chromosomes in the present study (Fig. 1G) suggests that they might have been incorporated into the Roma gene pool by admixture at different times in different geographical regions. Haplogroup R1a is particularly common in a large region extending from South Asia to Central Europe and Scandinavia (Underhill et al., 2010). R1a1-M198 was present in all the population groups, except the Tiszavasvari Roma. This lineage was absent from the Bulgarian and Iberian Roma as well (Gresham et al., 2001; Gusma˜o et al., 2008). Thus this lineage might have been introduced into their gene pool by host population admixture in the Carpathian Basin later, after fragmentation of the Roma groups from the Balkan regions. The network analysis of the Roma groups supports this possibility (Fig. 1H). Alternatively, some Roma groups might have lost this haplogroup by genetic drift. Haplogroup R1b is most common in Western Europe, and it is present at lower frequencies throughout Eastern Europe, Western Asia, and Central Asia (Cruciani et al., 2010). The R1b1-P25 haplogroup was present in all Roma and the Hungarian reference populations, but it was absent from the Indians. The network analysis showed a diverse pattern (Fig. 1I), which suggests that most of the R1b1-P25 chromosomes might have been incorporated into the Roma gene pool by different admixture events at different times. In a wider comparison of 41 populations, all Roma groups in the MDS analysis were affiliated with Balkan and Anatolian populations (see Fig. 3). This observation is consistent with the possibility that these areas provided an important geographic link between the Middle East, Asia and Europe during their genetic journey. The Hungarian reference population shows closer genetic affinity to the Upper Castes from India than to the Balkan populations, which may reflect the presence of a relatively high frequency of the R1a1 lineage in Hungarians as well as in the Indian Upper Castes (Sharma et al., 2009; Vo¨lgyi et al., 2009). This may support the hypothesis of the R1a1 haplogroup originating from common ancestors in Central Asia. The surprisingly low pairwise genetic distances (Rst) between Roma groups and Indians (see Fig. 4) supports the assumption of a common Indian origin as well. onlinelibrary.wiley.com/doi/10.1002/ajpa.21454/abstract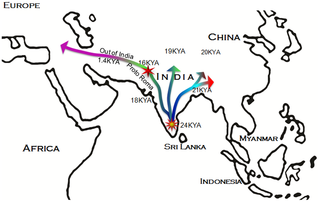 Figure 1. The most parsimonious route of prehistoric expansion of Y-chromosomal haplogroup H1a1a-M82 and the recent out-of -India migration of European Roma ancestors. Figure 1. The most parsimonious route of prehistoric expansion of Y-chromosomal haplogroup H1a1a-M82 and the recent out-of -India migration of European Roma ancestors.Although the Indian origin of the European Roma populations is linguistically and genetically well-established, accurate identification of their South Asian source has remained a matter of debate. Some linguistic studies argue that the proto-Romani founder population must have been in northwestern India [22]–[24], although their own origin myth suggests an origin on the Gangetic plain [25], [26]. The classical and mtDNA genetic markers suggested the closest affinity of the Roma with Rajput and Punjabi populations from Rajasthan and the Punjab respectively [16], [20], although these studies were compromised by low level phylogenetic resolution and a limited coverage of the Indian populations. These findings are now reconciled in light of our present genetic study thoroughly, comparing paternal lineages of Roma and a large number of South Asian populations, throwing light on the relationships and recent split of these groups. In contrast with the maternal lineages, where three South Asian-specific lineages were reported [16], the paternal lineage of Roma carries only a single currently discernible South Asian-specific Y-chromosomal founder hg H1a1a-M82, ranging from 10 to 60% frequency in various Roma populations [10], [17], [27]–[31]. The paternal ancestor of hg H1a1a-M82, viz. hg H-M69, is an autochthonous component of the South Asian Y-chromosome gene pool, accounting, on average, for 22% of the studied paternal lineage pools [32]–[36]. Its relatively deep phylogenetic position within the overall Y-chromosome phylogeny as well as its extremely low frequency elsewhere, including the East and Southeast Asia, Middle East and Europe [32], [37]–[43], suggests that it arose in South Asia sometime prior to the Last Glacial Maximum, perhaps even earlier between 30 and 40 KYA ago [33]. 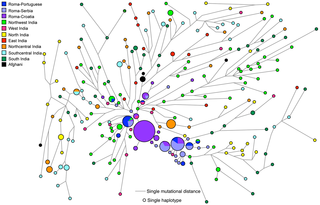 Figure 2. Phylogenetic network relating Y-STR haplotypes within haplogroup H1a1a -M82. Figure 2. Phylogenetic network relating Y-STR haplotypes within haplogroup H1a1a -M82.
The network was constructed using a median joining with MP (maximum parsimony) algorithm as implemented in the Network 4.6 program. The size of the circles is proportional to the number of samples. The data used for comparison have been taken from the literature (See Text S1).There are examples where a single Y-chromosomal hg is associated with the expansion of a population from a homeland, e.g. hg C3 with Genghis Khan's army [44], spread of hg O1-M110 bearing Austronesians from Taiwan to the Admiralty Islands of Melanesia [45] and hg O2a-M95 with the incursion of Austroasiatic speakers into India [35]. The reduced diversity and expansion of H1a1a-M82 lineages in all Roma groups argues for a descent from a single paternal ancestor in the Indian subcontinent [10], [29]. The most popular model proposes an origin in northwestern India, but this model has not been tested using an adequately large collection of data from Y-chromosomes of the Indian subcontinent. Here, we address this issue by generating the largest dataset to date of H1a1a-M82 lineages including 214 ethnic populations from India that fill a major geographical gap (Table S1). Hg H1a1a-M82 accounts for more than 12% of all male lineages in South Asia, with a highest incidence of 20% amongst South Indians and a lowest incidence of 0.2% among northeast Indian populations (Table S1 and S3). Hg H1a1a-M82 has a decreasing gradient from its peak frequency in southern India toward both the northwestern and the northeastern peripheries and a virtual absence further to the extreme east and west (Table S1, S3 and S4). Interestingly, substantial frequency changes occur despite the short geographical distance between the East Indian and their adjacent northeast Indian populations, suggesting different paternal population histories for these two neighboring regions. Consistent with the frequency distribution of H1a1a-M82 within India, the Y-STR variance and expansion ages basically reflect a similar pattern (Fig. 1 and Table 1). The overall TMRCA estimate of the H1a1a-M82 lineage in India using ρ-statistics [46], was ~22KYA. The regional expansion times within India range between 16 and 24 KYA (Table 1). The higher expansion time with the associated mean pair-wise difference (MPD) and haplotype diversity (HD) (Fig. 1 and, Table 1 and 2) suggests that hg H1a1a-M82 originated in the same South Asian pre-LGM gene pool of Y-chromosomes. Within India, language group-wise expansion times and variance were observed to be highest among Dravidian speakers (25±3.5 KYA, 0.59), followed by Austroasiatic speakers (22.4±3.3 KYA, 0.52), and lowest among Indo-European speakers (19.5±2.2 KYA, 0.47). Just as with the expansion time of hg O2a-M95 among Austroasiatic speakers [35], the H1a1a-M82 expansion time was established to be substantially higher among South Munda (22±3.5 KYA) than in the North Munda (16.3±3 KYA) group. The South Munda groups live in close proximity to the Dravidian groups, and it is likely that the already diverse Dravidian H1a1a-M82 might have assimilated into the Munda group after the Austroasiatic incursion into the Indian subcontinent. The geographical distribution of hg H1a1a-M82 is largely restricted to South Asia, and its significant occurrence among European Roma populations strongly links the Roma to the Indian subcontinent. The high frequency of H1a1a-M82 among all the Roma groups and their reduced genetic diversity relative to South Asian populations can most likely be attributed to their recent migration from India (Table 1 and 2). In the network analysis (Fig. 2), the Roma Y-STR haplotypes cluster predominantly close to the northwestern Indian haplotypes. In addition, the northwestern Indian haplotypes, while diverse, generally radiate from the core of the network while the Roma haplotypes being distributed further away. These patterns point to northwestern India as the source of the Roma H1a1a-M82 chromosomes. The average age estimate of Roma founders considering their distance from Northwest Indian founders is 1405±688 YBP (Table 3), which is largely in agreement with the time frames suggested previously. www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0048477 |
|
|
|
Post by Admin on Oct 30, 2013 21:33:33 GMT
 This image shows diversity in premolar and molar morphology in Neanderthals, modern humans and potential ancestral species. The identity of the last common ancestor of Neanderthals and modern humans is a controversial issue. This debate has been often addressed by means of descriptive analyses that are difficult to test. Our primary aim is to put questions about human evolution into a testable quantitative framework and to offer an objective means to sort out apparently unsolvable debates about hominin phylogeny. Our paper shows that no known hominin species matches the expected morphology of this common ancestor. Furthermore, we found that European representatives of potential ancestral species have had affinities with Neanderthals for almost 1 My, thus supporting a model of early divergence between Neanderthals and modern humans. A central problem in paleoanthropology is the identity of the last common ancestor of Neanderthals and modern humans ([N-MH]LCA). Recently developed analytical techniques now allow this problem to be addressed using a probabilistic morphological framework. This study provides a quantitative reconstruction of the expected dental morphology of the [N-MH]LCA and an assessment of whether known fossil species are compatible with this ancestral position. We show that no known fossil species is a suitable candidate for being the [N-MH]LCA and that all late Early and Middle Pleistocene taxa from Europe have Neanderthal dental affinities, pointing to the existence of a European clade originated around 1 Ma. These results are incongruent with younger molecular divergence estimates and suggest at least one of the following must be true: (i) European fossils and the [N-MH]LCA selectively retained primitive dental traits; (ii) molecular estimates of the divergence between Neanderthals and modern humans are underestimated; or (iii) phenotypic divergence and speciation between both species were decoupled such that phenotypic differentiation, at least in dental morphology, predated speciation. The missing ancestor ought to have teeth that look a bit more like the choppers of Homo ergaster, which wandered around eastern and southern Africa 1.3 to 1.8 million years ago, according to the study. “We can rule out all the European species as possible ancestors, because they are already in the line leading to Neanderthals, and we have an African species, which is the most similar one to the ancestor morphology,” Gomez-Robles said. “So the most intuitive explanation is that some African species posterior to Homo ergastor will be the ancestor.” The study suggested that ages derived from DNA modeling were underestimated. Alternatively, teeth may have started changing more rapidly for modern humans, or tooth shape was changing before such species diverged. Neither of those hypotheses is supported by the data or by commonly accepted models of evolution, Gomez-Robles noted. www.pnas.org/content/early/2013/10/15/1302653110.abstract?sid=3f308e69-91d8-4e90-993b-25f33078b8b2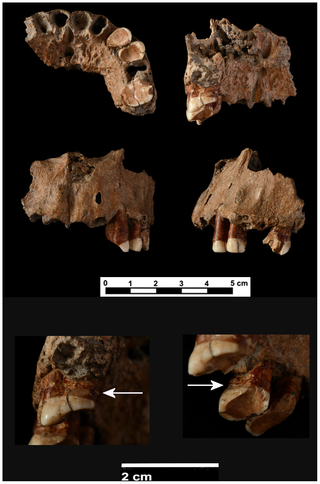 Different general views (inferior, internal, frontal and left) of the maxilla from Cova Foradà site. Detailed images of the last row: lower left, the arrow shows the interproximal groove on distal surface of left M1. Lower right, the arrow shows the interproximal groove on distal surface of left Pm3. Different general views (inferior, internal, frontal and left) of the maxilla from Cova Foradà site. Detailed images of the last row: lower left, the arrow shows the interproximal groove on distal surface of left M1. Lower right, the arrow shows the interproximal groove on distal surface of left Pm3.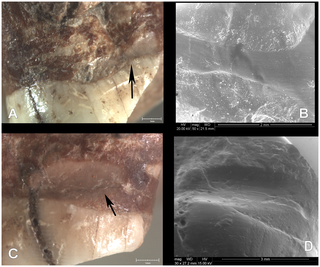 A: Interproximal groove on distal surface of left Pm3 (binocular lens image). B: Detailed view of the interproximal groove on Pm3 (ESEM image, 100×). C: Interproximal groove on distal surface of left M1 (binocular lens image). D. Detailed view of the interproximal groove on M1 (ESEM image, 30×). A: Interproximal groove on distal surface of left Pm3 (binocular lens image). B: Detailed view of the interproximal groove on Pm3 (ESEM image, 100×). C: Interproximal groove on distal surface of left M1 (binocular lens image). D. Detailed view of the interproximal groove on M1 (ESEM image, 30×).Interproximal grooves were documented on the distal surfaces of the Pm3 and M1. They were caused by the action of pulling some type of hard, narrow object, such as a toothpick, between adjacent teeth. Both interproximal grooves are related to heavy dental wear and periodontal disease. The adjacent teeth, the left Pm4 and upper left M2, were lost postmortem because there is no evidence of new bone formation in either alveoli. We do not know if those teeth suffered any dental pathology such as dental decay. However, the compensatory eruption of teeth with root exposition as a consequence of heavy dental wear could facilitate the introduction of food detritus between the teeth causing irritation and pain. The use of a small piece of wood or bone as a toothpick would alleviate that discomfort. The habit of picking the teeth causes interproximal grooves such as those found in an experimental study using grass stalks [18]. Interproximal grooves have been widely documented on the teeth of fossil populations (Table 1). A lower molar of Homo erectus (OH60) from Olduvai dated to 1.84 m.a. and the upper premolar of L894-I from Omo dated to 1.8 m.a. present the earliest evidence of these grooves [4], [18]. This feature is broadly documented in other hominins such as Homo heidelbergensis from the Sima de los Huesos site (Sierra de Atapuerca, Spain) and in Neanderthals [1], [19], [20], [21], [22], [23], [24] (Table 1). In our own species, Homo sapiens, interproximal grooves have been documented from Upper Palaeolithic individuals to present day aboriginal populations [20], [25], [26], [27], [28] (Table 1). In all populations and individuals, groove morphology is characterised by a tubular cross-section and labiolingual orientation. Interproximal grooving is more common on molars and premolars than on anterior teeth. There is no clear preference for mesial or distal surfaces, and there are many examples with both surfaces affected (Table 1). It is always caused by the introduction of some type of hard, thin and rigid probe between the teeth. Interproximal grooves on healthy teeth can be caused by using a pick between the teeth in order to remove food particles [2], [3], [22], [29], [30], [31], [32], [33]. Another aetiology for grooving on anterior and posterior teeth is linked to the use of the teeth as tools [27]. However, when grooves are documented on teeth with dental decay, heavy dental wear and periodontal disease, they could be the result of attempts to mitigate the inflammation of the gums [26], [28]. The interproximal grooves in CF-1 may be related to this last aetiology because both grooves are associated with periodontal disease and severe dental wear. In the last years, the evidence of the complex cognitive and behavioural capabilities of the Neanderthals has been increased. It has been proved that Neanderthals could speak and they were right-handed in the same proportion of modern people [34], [35], [36]. Neanderthals had a complex cultural organisation with an important symbolic behaviour such as burials or the use of feathers and claws as a personal garment demonstrate [37], [38], [39]. They also had thorough knowledge of the natural resources of their environment. Neanderthals from El Sidrón Cave (Spain) developed the ability to use medicinal plants, so they had some knowledge of medical treatment [40]. The use of toothpicks of plant origin to mitigate sore gums could also be considered as a type of rudimentary dental treatment. In sum, the use of toothpicks can be considered one of the most ancient habits documented in our genus, Homo. Sometimes, this habit may be related to a primitive form of oral hygiene to remove food particles. But, if interproximal grooves are associated with a dental pathology such as that suffered by the Cova Foradà specimen, the habit of using a tool to pick the teeth may be considered early evidence of medical treatment to alleviate sore gums. www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0076852?utm_source=feedburner&utm_medium=feed&utm_campaign=Feed%3A+plosone%2FPLoSONE+(PLOS+ONE+Alerts%3A+New+Articles) |
|
|
|
Post by Admin on Nov 15, 2013 6:08:42 GMT
 Population geneticists have formulated three hypotheses to explain the greater genetic diversity of Southern Europeans. One says that about 20,000 years ago, people from all over Europe retreated southward to escape advancing glaciers. When the glaciers receded, only a small segment of the original population recolonized the North. A second explanation, associated with the Neolithic revolution and the development of agriculture in the Near East, about 10,000 years ago, is that people from the Near East migrated westward to Europe, with gene flow varying in different parts of the continent. The third hypothesis says that Southern Europe's high genetic diversity was caused by recent migration from Africa to Southern Europe, related to the Roman conquest of North Africa and the Moorish occupation of the Iberian peninsula. However, previous studies suggested that Europeans and Africans share only between 1 and 3 percent of their genomes, making this scenario unlikely.  These studies looked at populations from sub-Saharan Africa but excluded North African populations. To correct this oversight, Bustamante and his team analyzed data from DNA samples of 2,099 individuals in 43 different populations, including seven from North Africa and three from Spain. They found much higher levels of gene flow from Africa to Europe than earlier studies had indicated. Evidence of North African descent was highest in the Iberian Peninsula and the Canary Islands, with up to 20 percent of individual genomes reflecting shared ancestry. The analysis showed that North Africans must have migrated to Spain at least 240 to 300 years ago. Basques who live on the Iberian Peninsula were an exception. They showed lower levels of shared ancestry with North Africans, closer to those of Europeans from further north. Human genetic diversity in southern Europe is higher than in other regions of the continent. This difference has been attributed to postglacial expansions, the demic diffusion of agriculture from the Near East, and gene flow from Africa. Using SNP data from 2,099 individuals in 43 populations, we show that estimates of recent shared ancestry between Europe and Africa are substantially increased when gene flow from North Africans, rather than Sub-Saharan Africans, is considered. The gradient of North African ancestry accounts for previous observations of low levels of sharing with Sub-Saharan Africa and is independent of recent gene flow from the Near East. The source of genetic diversity in southern Europe has important biomedical implications; we find that most disease risk alleles from genome-wide association studies follow expected patterns of divergence between Europe and North Africa, with the principal exception of multiple sclerosis.  Another possible hypothesis to explain the increased diversity in southern Europe is that an influx of Jewish ancestry had a heterogeneous effect on genetic diversity in Europe. However, in most European populations here, virtually no Jewish ancestry was detected. On average, 1% of Jewish ancestry is found in Tuscan HapMap population and Italian Swiss, as well as Greeks and Cypriots. This may reflect the higher sharing with Near Eastern populations in the Italian peninsula and southeastern Europe (Fig. 2C) or low levels of gene flow with the early Italian Jewish communities (6). Estimates from the IBD analysis are in agreement with ADMIXTURE estimates that the amount of sharing between these populations is extremely low (SI Appendix, Table S3). Specifically, results of IBD sharing between southwestern Europe and North Africa are two orders of magnitude greater than those found between the same region and Jews, the average WEA for southern Europe and North Africa is 203, while for southwestern Europe and European Jews is 1.3. Botigué, Laura R., et al. "Gene flow from North Africa contributes to differential human genetic diversity in southern Europe." Proceedings of the National Academy of Sciences (2013).Southern Europeans and Middle Eastern populations are known to have inherited a small percentage of their genetic material from recent sub-Saharan African migrations, but there has been no estimate of the exact proportion of this gene flow, or of its date. Here, we apply genomic methods to show that the proportion of African ancestry in many Southern European groups is 1%–3%, in Middle Eastern groups is 4%–15%, and in Jewish groups is 3%–5%. To estimate the dates when the mixture occurred, we develop a novel method that estimates the size of chromosomal segments of distinct ancestry in individuals of mixed ancestry. We verify using computer simulations that the method produces useful estimates of population mixture dates up to 300 generations in the past. By applying the method to West Eurasians, we show that the dates in Southern Europeans are consistent with events during the Roman Empire and subsequent Arab migrations. The dates in the Jewish groups are older, consistent with events in classical or biblical times that may have occurred in the shared history of Jewish populations.  Table 2. Estimates of mixture proportions and date of mixture. Table 2. Estimates of mixture proportions and date of mixture.Application of f4 Ancestry Estimation suggests that the highest proportion of African ancestry in Europe is in Iberia (Portugal 3.2±0.3% and Spain 2.4±0.3%), consistent with inferences based on mitochondrial DNA [6] and Y chromosomes [7] and the observation by Auton et al. [8] that within Europe, the Southwestern Europeans have the highest haplotype-sharing with Africans. The proportion decreases to the north and we find no evidence for mixture in Russia, Sweden and Scotland (Table 2, Figure S5). We also detect about 3-5% sub-African ancestry in all the Jewish populations, a finding that is novel as far as we are aware, and certainly has not been unambiguously demonstrated or quantified. For Levantines, the proportions are often higher: 9.3%±0.4% in Palestinians and >10% in the Bedouins (standard errors were calculated using a Block Jackknife as described in Materials and Methods). Table 2 presents the ancestry estimates that we obtain for all West Eurasian populations with significant evidence of mixture by the 4 Population Test (Z-score < -3). To test if our inferences are dependent on the sub-Saharan African population that was used as the reference group, we also repeated analyses with other sub-Saharan African populations replacing YRI. This analysis shows that our estimates of mixture proportions do not change significantly based on the ancestral population used (Text S2c, Table S6). We obtained similar estimates when we applied STRUCTURE 2.2 [22] to estimate the mixture proportions using ~13,900 independent markers (that were not in linkage disequilibrium (LD) with each other) (Table 2, Figure S6).  Figure 3. Testing for LD due to African admixture in West Eurasians. Figure 3. Testing for LD due to African admixture in West Eurasians.
To generate these plots, we used the ROLLOFF software to calculate the LD between all pairs of markers in each population, weighted by their frequency difference between YRI and CEU to make the statistic sensitive to admixture LD. We plot the correlation as a function of genetic distance for Portuguese, Russians, Sephardic Greek Jews and Palestinians. We do not show inter-SNP intervals of <0.5cM since we have found that at this distance admixture LD begins to be confounded by background LD, and so inferences are not reliable (exponential curve fitting does not include inter-SNP intervals at this scale).The finding of African ancestry in Southern Europe dating to ~55 generations ago, or ~1,600 years ago assuming 29 years per generation [30], needs to be placed in historical context. The historical record documents multiple interactions of African and European populations over this period. One potential opportunity for African gene flow was during the period of Roman occupation of North Africa that lasted until the early 5th century AD, and indeed tomb inscriptions and literary references suggest that trade relations continued even after that time [31], [32]. North Africa was also a supplier of goods and products such as wine and olive oil to Italy, Spain and Gaul from 200–600 AD, and Morocco was a major manufacturer of the processed fish sauce condiment, garum, which was imported by Romans [33]. In addition, there was slave trading across the western Sahara during Roman times [7], [34]. Another potential source of some of the African ancestry, especially in Spain and Portugal, is the invasion of Iberia by Moorish armies after 711 AD [35], [36]. If the Moors already had some African ancestry when they arrived in Southern Europe, and then admixed with Iberians, we would expect the admixture date to be older than the date of the invasion, as we observe. The signal of African mixture that we detect in Levantines (Bedouins, Palestinians and Druze) – an average of 32 generations or ~1000 years ago – is more recent than the signal in Europeans, which might be related to the migrations between North Africa and Middle East that have occurred over the last thousand years, and the proximity of Levantine groups geographically to Africa. Syria and Palestine were under Egyptian political control until the 16th century AD when they were conquered by the Ottoman Empire. This is in concordance with our proposed dates. In addition, the Arab slave trade is responsible for the movement of large numbers of people from Africa across the Red Sea to Arabia from 650 to 1900 AD and probably even prior to the Islamic times [7], [37]. We caution that our sampling of the Middle East is sparse, and it will be of interest to study African ancestry in additional groups from this region. A striking finding from our study is the consistent detection of 3–5% sub-Saharan African ancestry in the 8 diverse Jewish groups we studied, Ashkenazis (from northern Europe), Sephardis (from Italy, Turkey and Greece), and Mizrahis (from Syria, Iran and Iraq). This pattern has not been detected in previous analyses of mitochondrial DNA and Y chromosome data [7], and although it can be seen when re-examining published results of STRUCTURE-like analyses of autosomal data, it was not highlighted in those studies, or shown to unambiguously reflect sub-Saharan African admixture [15], [38]. www.plosgenetics.org/article/info%3Adoi%2F10.1371%2Fjournal.pgen.1001373 |
|
|
|
Post by Admin on Nov 27, 2013 22:57:34 GMT
A genetic study (Friedlaender et al. 2007) found the spotty distribution of the African haplogroup E1b in New Britain (the Papuan-speaking Ata and Sulka) and across a number of Oceanic speaking groups in the region, showing that some of those Melanesians are partially of African descent. It's been speculated that ancient migrants of African decent had reached Latin America after ancient skulls and artifacts in South America that looked more like those belonging to indigenous Australians and Melanesians than Northern Asians were discovered. It's unlikely that they had crossed the Bering Strait prior to North Asian migrations and they may have migrated to Latin America at least 3,000 years earlier by taking sea routes as there is no evidence of Melanesian colonisation in North America. But ancient African migrations have altered the genetic make-up of Melanesians significantly and haplogroup E1b was also found in other Asian countries such as Thailand and Taiwan.  Olmec head or colossal head labeled as number 1 in the Xalapa's museum of Antropology. It is also known as el rey (the king) It was found in San Lorenzo Tenochtitlán (name of the archeological site, usually shortened to San Lorenzo), located at Texistepec, State of Veracruz, México. It dates from 1200 to 900 years b.C. and is 2.9 meters high and 2.1 meters wide. Olmec head or colossal head labeled as number 1 in the Xalapa's museum of Antropology. It is also known as el rey (the king) It was found in San Lorenzo Tenochtitlán (name of the archeological site, usually shortened to San Lorenzo), located at Texistepec, State of Veracruz, México. It dates from 1200 to 900 years b.C. and is 2.9 meters high and 2.1 meters wide.Melanesian populations are known for their diversity, but it has been hard to grasp the pattern of the variation or its underlying dynamic. Using 1,223 mitochondrial DNA (mtDNA) sequences from hypervariable regions 1 and 2 (HVR1 and HVR2) from 32 populations, we found the among-group variation is structured by island, island size, and also by language affiliation. The more isolated inland Papuan-speaking groups on the largest islands have the greatest distinctions, while shore dwelling populations are considerably less diverse (at the same time, within-group haplotype diversity is less in the most isolated groups). Persistent differences between shore and inland groups in effective population sizes and marital migration rates probably cause these differences. We also add 16 whole sequences to the Melanesian mtDNA phylogenies. We identify the likely origins of a number of the haplogroups and ancient branches in specific islands, point to some ancient mtDNA connections between Near Oceania and Australia, and show additional Holocene connections between Island Southeast Asia/Taiwan and Island Melanesia with branches of haplogroup E. Coalescence estimates based on synonymous transitions in the coding region suggest an initial settlement and expansion in the region at ~30–50,000 years before present (YBP), and a second important expansion from Island Southeast Asia/Taiwan during the interval ~3,500–8,000 YBP. However, there are some important variance components in molecular dating that have been overlooked, and the specific nature of ancestral (maternal) Austronesian influence in this region remains unresolved. 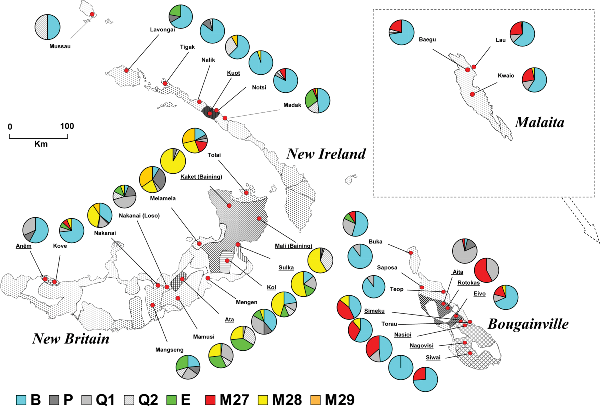 The haplogroup variation in the core region of Northern Island Melanesia (table S2 shows the actual haplogroup incidences). While there is an island-by-island distinction, New Britain is considerably more internally diverse than Bougainville, with both New Ireland and Malaita considerably less so. The haplogroup variation in the core region of Northern Island Melanesia (table S2 shows the actual haplogroup incidences). While there is an island-by-island distinction, New Britain is considerably more internally diverse than Bougainville, with both New Ireland and Malaita considerably less so.Haplogroup E. This relatively uncommon haplogroup, a subdivision of M9, was thought to be limited to Mainland and Island Southeast Asia [45]. Its E1a branch had been found in Thailand, in Sabah Aborigines, in Taiwan Aborigines, as well as across Indonesia. The E1b branch had been found in Indonesia and the Philippines (apparently absent in Taiwan). The rarer E2 had only been found in Taiwan Aborigines and Filipinos. We have added 5 complete E sequences now identified in Northern Island Melanesia (figure 5). The Island Melanesian E1a branches share a key mutation with one from the Philippines (nts 373), and the Melanesian E1b branches share 4 mutations with another branch from the Philippines.
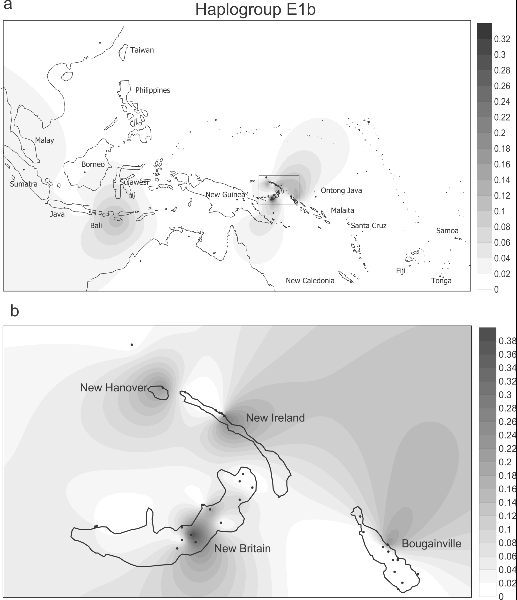
Spatial frequency distribution of haplogroup E1b in Island Southeast Asia and the western Pacific (7a), and details for Northern Island Melanesia (7b). The distribution of E in our series was spotty (table S2). 75 samples were E1b, and most of these were from New Britain (the Papuan-speaking Ata and Sulka). The rest were scattered across a number of Oceanic speaking groups in the region. We have not been able to ascertain from the literature if E was dispersed to Remote Oceania, since its identification depends on sequencing nts 16390 in HVS1. Figures 6 and 7 give a sense of the heterogeneous distributions of E1a and E1b in Island Melanesia and the Southwest Pacific as currently understood (table S3 has the data references). We also found two E2s, which have been identified in Taiwan as well as Indonesia and the Philippines. In sum, E is a second young haplogroup that was brought to Near Oceania, specifically to New Britain, from the west. Coalescence time estimates for E1a and E1b in our series suggest both branches are about the same age as the “Polynesian Motif,” or slightly younger (table 1). The distribution of E1b suggests a connection between Island Southeast Asia (excluding Taiwan) and Northern Island Melanesia. The distributions in Near Oceania of the haplogroups B4b1, F, M7, and Y are too rare to be informative. PLOS ONE: Melanesian mtDNA Complexity
 Frequency distribution of (A, B) NRY and (C, D) mtDNA haplogroups found in Polynesia with a genetic origin in (A, C) Asia or (B, D) Melanesia. Frequency distribution of (A, B) NRY and (C, D) mtDNA haplogroups found in Polynesia with a genetic origin in (A, C) Asia or (B, D) Melanesia.The colonization of Polynesia which ranges from Hawaii in the north to Easter Islands in the east, Fiji in the west, and New Zealand in the south, is still a matter of debate. According to linguistic evidence, Polynesian languages are closely related to each other and belong to the Austronesian language family that can be traced back to East Asia, in particular to the present-day languages of Taiwanese Aborigines (Blust 1999; Diamond 2000). Furthermore, linguistic evidence (Gray and Jordan 2000) is usually interpreted to support the “Express-train” hypothesis (Diamond 1988), according to which Polynesian ancestors moved rapidly from Eastern Asia into the Pacific without significant admixture with Melanesians (we use the term “Melanesia” in the geographic sense, to include here the mainland of New Guinea and surrounding islands, also referred to as Near Oceania). Archaeological evidence suggests that western Polynesian islands (Fiji, Futuna, Samoa, Tonga) were settled 2,100–3,200 years ago by people belonging to the so-called Lapita cultural complex that originated 3,000–3,500 years ago in Island Melanesia, in particular the Bismarck Archipelago (Kirch 2000). However, some archaeologists argue that the Lapita cultural complex originated about 6,000 years ago in China and thus associate the spread of Austronesian languages with the Neolithic spread of material culture, including agriculture and Lapita, from East Asia into the Pacific under the Express-train scenario (Bellwood 1978; Diamond and Bellwood 2003), whereas others suggest a strict Melanesian origin of the Lapita cultural complex (White et al. 1988; Terrell 1989; Terrell et al. 2001). Besides the 2 “extreme” models, the “Express train” assuming an Asian origin of Polynesians with no or little admixture of ingenious Melanesians and the “Entangled bank” assuming a long and complex history of human interactions starting from the first occupation of Melanesia in the Pleistocene (Terrell 1988), there are additional “intermediate” models such as the “Triple I” (Green 1991). The Triple I model assumes that components of the Lapita cultural complex are results of Intrusions of nonindigenous Asian components together with the Integration of indigenous Melanesian elements and new Innovations (Green 1991). In contrast to the clear evidence for an Asian origin of Polynesian languages and a probable Melanesian origin of the Lapita material culture found in Polynesia, the genetic origin of Polynesians is still contentious. Studies of maternally inherited mtDNA markers have favored an Asian origin of Polynesian maternal lineages (Melton et al. 1995; Redd et al. 1995; Sykes et al. 1995; Trejaut et al. 2005) in support of the Express-train hypothesis. In contrast, studies of paternally inherited DNA markers from the nonrecombining portion of the Y chromosome (NRY) have revealed a mostly Melanesian origin of Polynesian paternal lineages (Kayser, Brauer et al. 2000; Capelli et al. 2001; Underhill, Passarino, Lin, Marzuki et al. 2001; Hurles et al. 2002) supporting the “Slow Boat” hypothesis (Kayser, Brauer et al. 2000). The Slow Boat model assumes that Polynesian ancestors originated in Eastern Asia but mixed extensively with indigenous Melanesians before colonizing the Pacific (Kayser, Brauer et al. 2000). Unfortunately, a similar term “Slow boat to Melanesia” was subsequently used to suggest a Southeast Asian genetic origin of Polynesians in the Pleistocene based on mitochondrial DNA (mtDNA) evidence (Diamond 2001; Oppenheimer and Richards 2001). Studies of autosomal DNA markers suggest different scenarios depending on the markers used, for example, a Melanesian origin of Polynesian hemoglobin genes (Hill et al. 1985, 1987) versus an Asian origin of Polynesian human leucocyte antigen (HLA) genes (Mack et al. 2000; Mack and Erlich 2005).  Two-dimensional plots from multidimensional scaling analyses based on FST distances from (A) NRY haplogroup frequencies and (B) mtDNA sequence haplotypes. NRY haplogroups R-M173 and F-M89 were omitted due to their assumed European origin; Niue (NRY) and Tokelau (NRY, mtDNA) were omitted due to small sample size. For population abbreviations, see Table S5(Supplementary Material online). Geographic regions are highlighted. Two-dimensional plots from multidimensional scaling analyses based on FST distances from (A) NRY haplogroup frequencies and (B) mtDNA sequence haplotypes. NRY haplogroups R-M173 and F-M89 were omitted due to their assumed European origin; Niue (NRY) and Tokelau (NRY, mtDNA) were omitted due to small sample size. For population abbreviations, see Table S5(Supplementary Material online). Geographic regions are highlighted.These results indicate the central role of Fiji in further Polynesian migrations; the fact that Fiji has the highest genetic diversity, and that all Polynesian groups have a subset of the diversity in Fiji, indicates that humans probably first migrated to Fiji and that subsequent settlement of Polynesia probably came from Fiji. This is in agreement with archaeological evidence showing that the oldest findings of Lapita pottery in Polynesia are from Fiji (3,200 years ago). Having originated from the Bismark Archipelago in Island Melanesia, Lapita was first introduced to Polynesia in Fiji, and there was a rapid expansion of the Lapita cultural complex from Fiji eastward into other parts of Polynesia (Futuna, Tonga, Samoa) as suggested indirectly by finding younger Lapita dates elsewhere in Polynesia (2,900–2,100 years ago), but also directly, for example, by the presence of Fijian potsherds in Tonga (Kirch 2000). An alternative explanation is that following initial colonization, Fiji continued to receive migrants and genes from Melanesia and that humans continued to disperse from Fiji to Polynesia. Although there is archaeological evidence to support this view (for summary see Kirch 2000), the genetic results do not suggest substantial ongoing contact between Fiji and Melanesia, as separate expansions of Y haplogroups C-M208 and K-M9 (both of Melanesian origin) in Fiji/Polynesia versus Melanesia are evident in the networks (fig. 2A and B). Ongoing contact between Melanesia and Fiji should result in more sharing of haplotypes between Melanesia and Fiji, which is not observed. Moreover, Y haplogroup M-M4 (of Melanesian origin) has its highest frequency in Fiji and exists in Melanesia mostly as its derived subgroup M-P34; ongoing contact should have brought more M-P34 chromosomes to Fiji. However, the low frequency elsewhere in Polynesia of other Melanesian Y and mtDNA haplogroups existing in Fiji precludes definitive conclusions, and additional sampling between mainland New Guinea and Fiji (e.g., from the Solomon Islands, Vanuatu, and New Caledonia) is needed to further investigate the amount of ongoing genetic contact between Melanesia and Fiji. Kayser, Manfred, et al. " Melanesian and Asian origins of Polynesians: mtDNA and Y chromosome gradients across the Pacific." Molecular biology and evolution 23.11 (2006): 2234-2244. |
|

