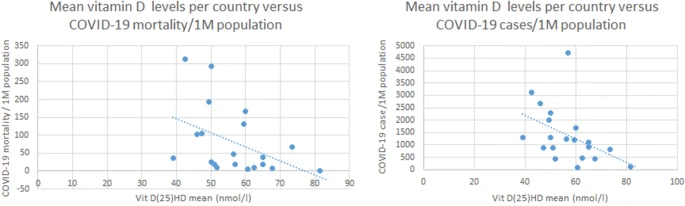
Post by Admin on May 1, 2020 22:07:29 GMT
ACE2 and acute lung injury
Experimental SARS-CoV infection induces acute respiratory failure and lung parenchymal injury characterized by alveolar wall thickening, pulmonary vascular hyperpermeability, and inflammatory cell infiltration in mice [27]. After SARS-CoV infection in mice, lung ACE2 protein levels are greatly reduced, while ACE levels are not changed [27]. These findings are consistent with the previous observation that coronaviruses specifically downregulate ACE2 expression in host cells, depending on virus replication [29]. In addition, SARS-CoV-induced acute lung injury is remarkably attenuated in ACE2 knockout mice compared with wild-type mice [27]. Lung angiotensin II levels increase in wild-type mice after SARS-CoV infection [27]. Acid aspiration mice, an ARDS model, show functional and pathological changes mimicking SARS-CoV-induced lung injury [30]. The lung angiotensin II levels increase after acid aspiration, and SARS-CoV-S protein treatment elicits a further angiotensin II increase [27]. Interestingly, exogenous recombinant ACE2 treatment rescues acid aspiration-induced acute lung injury [30]. Taking this evidence together, the following hypothesis has been raised for the mechanism underlying SARS-CoV-induced lung injury: SARS-CoV infection downregulates lung ACE2 and in turn, shifts the balance toward the dominance of the ACE/angiotensin II/AT1R system over the ACE2/angiotensin 1-7/mas receptor system in the lung. As a result, noncompeting angiotensin II accumulation occurs, resulting in acute lung injury through AT1R activation [27].
It should be noted that a similar mechanism is proposed for the severe lung injury caused by the avian influenza A virus H5N1, which has spread worldwide in humans with a high mortality rate. After H5N1 virus infection in mice, lung ACE2 expression is downregulated, and serum angiotensin II levels increase [31]. Acute lung injury is augmented by ACE2 knockout in H5N1-infected mice, while the administration of recombinant human ACE2 ameliorates H5N1-induced lung injury in mice [31]. Moreover, in other mouse models of acute lung injury/ARDS, such as acid aspiration, sepsis, and drug-induced lung fibrosis, it has been shown that ACE2 expression is downregulated in response to noxious stimuli and that acute lung injury is aggravated by ACE2 knockout and rescued by exogenous ACE2 administration or AT1R knockout [30, 32]. Accordingly, it is suggested that the angiotensin II/AT1R-induced aggravation resulting from ACE2 downregulation and the protective effects of ACE2 are not specific to lung injury in SARS but rather may be common to acute lung injury/ARDS induced by various viral infections and lung diseases.
Effects of RAS inhibitors on ACE2 and SARS-CoV-induced lung injury
Several kinds of ARBs (e.g., olmesartan, telmisartan, losartan, and azilsartan) have been shown to increase the mRNA or protein levels of ACE2 in animal models of heart diseases (hypertensive hypertrophy, autoimmune myocarditis, dilated cardiomyopathy, myocardial infarction, and diabetic cardiomyopathy) [8,9,10,11,12,13,14] and chronic kidney disease (hypertensive nephropathy and diabetic nephropathy) [15, 16], as well as normal rat heart and renal vasculature [17, 18] and hypertensive rat aorta (but not the carotid artery) [19, 20]. ACEI does not inhibit ACE2 as a pharmacological property. In normal rats, lisinopril has been shown to modestly increase cardiac ACE2 mRNA levels while not changing cardiac ACE2 activity [17]. In the myocardial infarction model, valsartan, ramipril, and their combination have no effects on cardiac ACE2 expression [33]. It should be noted that the doses of ARBs and ACEIs used in these animal studies were much greater than those in clinical practice so that the observed effects of these drugs on ACE2 expression and activity could not be extrapolated to clinical situations in humans. Importantly, no clinical data exist regarding the effects of ARBs and ACEIs on human tissue ACE2 expression or activity in vivo. Although there are several clinical studies investigating the changes in plasma or urine levels of the soluble form of ACE2 in hypertension and CVD patients receiving ARBs or ACEIs, the soluble form of ACE2 in the plasma and urine is not a reliable indicator of the activity of tissue ACE2, namely, membrane-bound ACE2 [22]. Importantly, there are no data on the effects of ARBs and ACEIs on lung ACE2 expression either in animal models or humans. It is still unknown whether the changes in ACE2 levels (for example, ARB-induced upregulation) actually facilitate greater engagement and entry of SARS-CoV and other viruses, even in animal models.
On the other hand, it has been shown that pretreatment with losartan, an ARB, ameliorates acute pulmonary edema and lung injury in SARS-CoV-S protein-treated mice after acid aspiration (Fig. 2) [27]. Taken together with the lung angiotensin II level elevation [27], this evidence suggested that even after considering the speculation that viral load would be enhanced by ACE2 upregulation, the net effects of ARBs would be favorable to prevent SARS-CoV-induced acute lung injury in this model. In addition, there is another possibility that the ARB pretreatment-induced baseline ACE2 increase prior to SARS-CoV infection would result in higher ACE2 levels that remained after SARS-CoV-induced downregulation and consequently conferred protection against lung injury in this model [22, 34]. However, in interpreting the results of this study, it should be noted that the effects of losartan have been observed merely in mice treated with SARS-CoV-S protein in addition to acid aspiration but not in mice treated with SARS-CoV-S protein alone. Additionally, data on lung ACE2 levels before and after treatment with SARS-CoV-S protein and acid aspiration are lacking in mice pretreated with or without losartan.
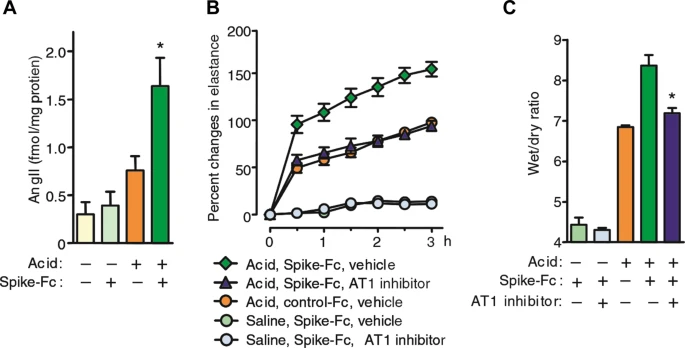
Fig. 2
Current evidence in COVID-19
Earlier studies reported that the case fatality rate was high in COVID-19 patients with comorbidities such as older age, hypertension, diabetes mellitus, CVD, chronic pulmonary disease, and malignancy [3, 4]. Because patients with hypertension and CVD are likely treated with ACEIs or ARBs, the concern is raised regarding whether the use of ACEIs and ARBs may aggravate the morbidity and mortality of COVID-19 [21]. However, it was possible that the observed phenomenon would result from reverse causality because older patients are at the highest risk for COVID-19 and concurrently tend to have multiple comorbidities, including hypertension and CVD, and because adjustments for age and other possible confounding factors were not performed in such earlier studies [22, 35, 36]. Indeed, in a recent retrospective, multicenter cohort study enrolling 191 confirmed COVID-19 patients in Wuhan, China, multivariable regression analysis revealed that independent risk factors for in-hospital death are older age, higher Sequential Organ Failure Assessment score, and higher D-dimer levels at the early stage of COVID-19, although hypertension, diabetes, and coronary artery disease were not included [37].
A small case study reported that plasma angiotensin II levels were markedly elevated and linearly associated with viral load and lung injury severity in COVID-19 pneumonia patients [38]. Given that a remarkable elevation of circulating levels was also documented in various kinds of cytokines (IL-6, IL-10, TNF-alpha, etc.), it is unknown whether these findings of angiotensin II are the cause or result of a systemic cytokine storm in COVID-19 patients [39]. In the context of the effects of ARBs and AECIs, a retrospective, single-center analysis enrolling 112 COVID-19 patients showed that the use of ARBs and ACEIs had no effects on the morbidity and mortality of COVID-19 patients with CVD [40]. Currently, a case-control study is underway to clarify the effects of ACEIs and ARBs on the severity of COVID-19 in Italy (NCT04318418). Most importantly, at present, there is no large-scale study with convincing evidence to determine whether ARBs and ACEIs play a neutral, beneficial, or harmful role in the susceptibility to SARS-CoV-2 and the severity and outcomes of COVID-19 patients [22, 34,35,36, 41].
Experimental SARS-CoV infection induces acute respiratory failure and lung parenchymal injury characterized by alveolar wall thickening, pulmonary vascular hyperpermeability, and inflammatory cell infiltration in mice [27]. After SARS-CoV infection in mice, lung ACE2 protein levels are greatly reduced, while ACE levels are not changed [27]. These findings are consistent with the previous observation that coronaviruses specifically downregulate ACE2 expression in host cells, depending on virus replication [29]. In addition, SARS-CoV-induced acute lung injury is remarkably attenuated in ACE2 knockout mice compared with wild-type mice [27]. Lung angiotensin II levels increase in wild-type mice after SARS-CoV infection [27]. Acid aspiration mice, an ARDS model, show functional and pathological changes mimicking SARS-CoV-induced lung injury [30]. The lung angiotensin II levels increase after acid aspiration, and SARS-CoV-S protein treatment elicits a further angiotensin II increase [27]. Interestingly, exogenous recombinant ACE2 treatment rescues acid aspiration-induced acute lung injury [30]. Taking this evidence together, the following hypothesis has been raised for the mechanism underlying SARS-CoV-induced lung injury: SARS-CoV infection downregulates lung ACE2 and in turn, shifts the balance toward the dominance of the ACE/angiotensin II/AT1R system over the ACE2/angiotensin 1-7/mas receptor system in the lung. As a result, noncompeting angiotensin II accumulation occurs, resulting in acute lung injury through AT1R activation [27].
It should be noted that a similar mechanism is proposed for the severe lung injury caused by the avian influenza A virus H5N1, which has spread worldwide in humans with a high mortality rate. After H5N1 virus infection in mice, lung ACE2 expression is downregulated, and serum angiotensin II levels increase [31]. Acute lung injury is augmented by ACE2 knockout in H5N1-infected mice, while the administration of recombinant human ACE2 ameliorates H5N1-induced lung injury in mice [31]. Moreover, in other mouse models of acute lung injury/ARDS, such as acid aspiration, sepsis, and drug-induced lung fibrosis, it has been shown that ACE2 expression is downregulated in response to noxious stimuli and that acute lung injury is aggravated by ACE2 knockout and rescued by exogenous ACE2 administration or AT1R knockout [30, 32]. Accordingly, it is suggested that the angiotensin II/AT1R-induced aggravation resulting from ACE2 downregulation and the protective effects of ACE2 are not specific to lung injury in SARS but rather may be common to acute lung injury/ARDS induced by various viral infections and lung diseases.
Effects of RAS inhibitors on ACE2 and SARS-CoV-induced lung injury
Several kinds of ARBs (e.g., olmesartan, telmisartan, losartan, and azilsartan) have been shown to increase the mRNA or protein levels of ACE2 in animal models of heart diseases (hypertensive hypertrophy, autoimmune myocarditis, dilated cardiomyopathy, myocardial infarction, and diabetic cardiomyopathy) [8,9,10,11,12,13,14] and chronic kidney disease (hypertensive nephropathy and diabetic nephropathy) [15, 16], as well as normal rat heart and renal vasculature [17, 18] and hypertensive rat aorta (but not the carotid artery) [19, 20]. ACEI does not inhibit ACE2 as a pharmacological property. In normal rats, lisinopril has been shown to modestly increase cardiac ACE2 mRNA levels while not changing cardiac ACE2 activity [17]. In the myocardial infarction model, valsartan, ramipril, and their combination have no effects on cardiac ACE2 expression [33]. It should be noted that the doses of ARBs and ACEIs used in these animal studies were much greater than those in clinical practice so that the observed effects of these drugs on ACE2 expression and activity could not be extrapolated to clinical situations in humans. Importantly, no clinical data exist regarding the effects of ARBs and ACEIs on human tissue ACE2 expression or activity in vivo. Although there are several clinical studies investigating the changes in plasma or urine levels of the soluble form of ACE2 in hypertension and CVD patients receiving ARBs or ACEIs, the soluble form of ACE2 in the plasma and urine is not a reliable indicator of the activity of tissue ACE2, namely, membrane-bound ACE2 [22]. Importantly, there are no data on the effects of ARBs and ACEIs on lung ACE2 expression either in animal models or humans. It is still unknown whether the changes in ACE2 levels (for example, ARB-induced upregulation) actually facilitate greater engagement and entry of SARS-CoV and other viruses, even in animal models.
On the other hand, it has been shown that pretreatment with losartan, an ARB, ameliorates acute pulmonary edema and lung injury in SARS-CoV-S protein-treated mice after acid aspiration (Fig. 2) [27]. Taken together with the lung angiotensin II level elevation [27], this evidence suggested that even after considering the speculation that viral load would be enhanced by ACE2 upregulation, the net effects of ARBs would be favorable to prevent SARS-CoV-induced acute lung injury in this model. In addition, there is another possibility that the ARB pretreatment-induced baseline ACE2 increase prior to SARS-CoV infection would result in higher ACE2 levels that remained after SARS-CoV-induced downregulation and consequently conferred protection against lung injury in this model [22, 34]. However, in interpreting the results of this study, it should be noted that the effects of losartan have been observed merely in mice treated with SARS-CoV-S protein in addition to acid aspiration but not in mice treated with SARS-CoV-S protein alone. Additionally, data on lung ACE2 levels before and after treatment with SARS-CoV-S protein and acid aspiration are lacking in mice pretreated with or without losartan.
Fig. 2
Current evidence in COVID-19
Earlier studies reported that the case fatality rate was high in COVID-19 patients with comorbidities such as older age, hypertension, diabetes mellitus, CVD, chronic pulmonary disease, and malignancy [3, 4]. Because patients with hypertension and CVD are likely treated with ACEIs or ARBs, the concern is raised regarding whether the use of ACEIs and ARBs may aggravate the morbidity and mortality of COVID-19 [21]. However, it was possible that the observed phenomenon would result from reverse causality because older patients are at the highest risk for COVID-19 and concurrently tend to have multiple comorbidities, including hypertension and CVD, and because adjustments for age and other possible confounding factors were not performed in such earlier studies [22, 35, 36]. Indeed, in a recent retrospective, multicenter cohort study enrolling 191 confirmed COVID-19 patients in Wuhan, China, multivariable regression analysis revealed that independent risk factors for in-hospital death are older age, higher Sequential Organ Failure Assessment score, and higher D-dimer levels at the early stage of COVID-19, although hypertension, diabetes, and coronary artery disease were not included [37].
A small case study reported that plasma angiotensin II levels were markedly elevated and linearly associated with viral load and lung injury severity in COVID-19 pneumonia patients [38]. Given that a remarkable elevation of circulating levels was also documented in various kinds of cytokines (IL-6, IL-10, TNF-alpha, etc.), it is unknown whether these findings of angiotensin II are the cause or result of a systemic cytokine storm in COVID-19 patients [39]. In the context of the effects of ARBs and AECIs, a retrospective, single-center analysis enrolling 112 COVID-19 patients showed that the use of ARBs and ACEIs had no effects on the morbidity and mortality of COVID-19 patients with CVD [40]. Currently, a case-control study is underway to clarify the effects of ACEIs and ARBs on the severity of COVID-19 in Italy (NCT04318418). Most importantly, at present, there is no large-scale study with convincing evidence to determine whether ARBs and ACEIs play a neutral, beneficial, or harmful role in the susceptibility to SARS-CoV-2 and the severity and outcomes of COVID-19 patients [22, 34,35,36, 41].