|
|
Post by Admin on May 24, 2020 7:42:30 GMT
The researchers started with archived samples of cells from the nasal lining, or epithelium, of people ages 4 to 60. Then they measured the activity of a gene that directs production of ACE2, a protein that helps coronavirus enter the body. It turned out that ACE2 gene “expression” — the DNA instructions that are converted into a functional molecule — was lower in young children, and that expression increased with age. “Few studies have examined the relationship between ACE2 in the airway and age," the team wrote in a letter published Wednesday in JAMA Network. "The results from this study show age-dependent expression of ACE2 in nasal epithelium, the first point of contact for [the coronavirus] and the human body." 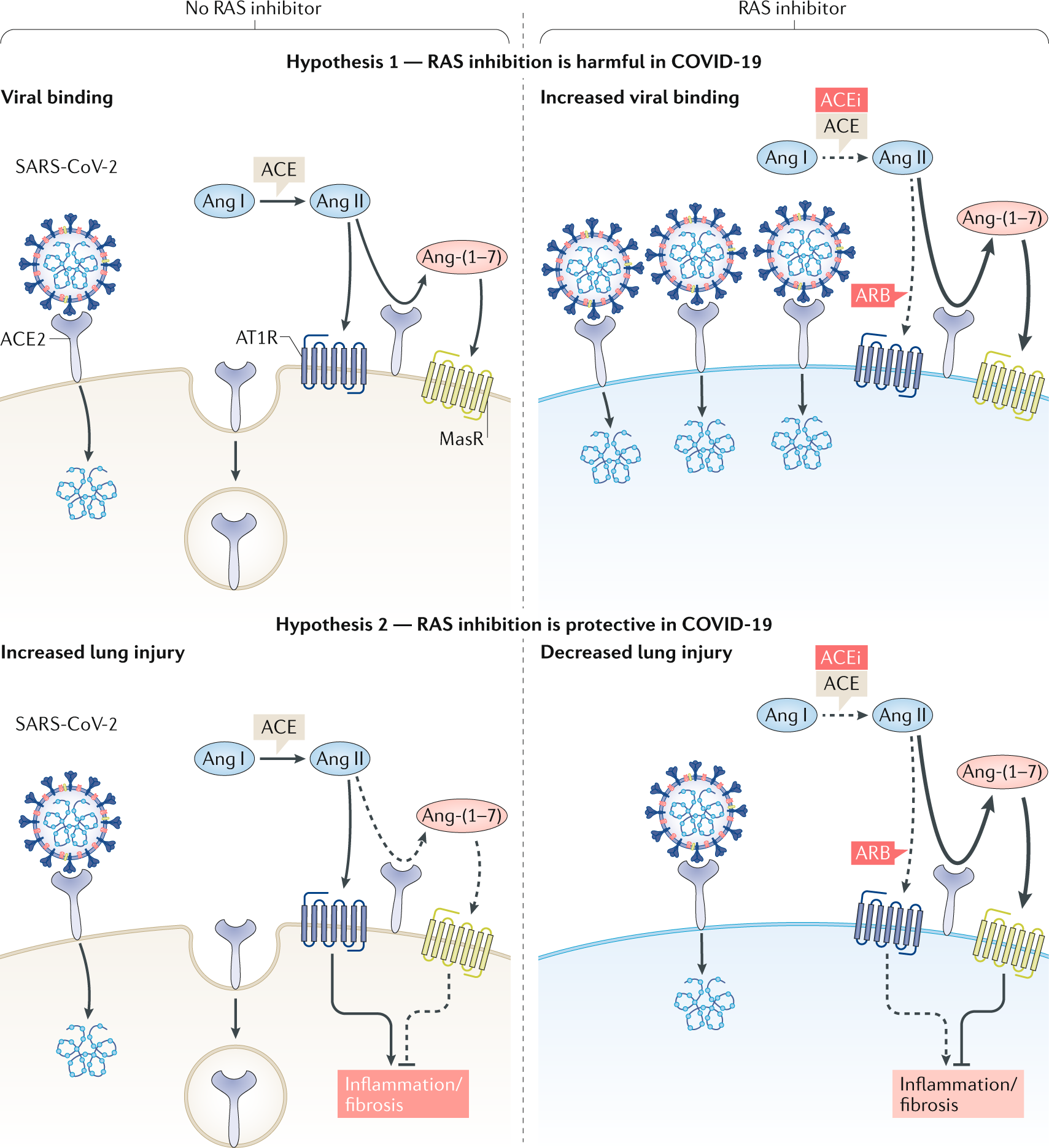 ] The implications of that are intriguing, but not yet clear, said Ishminder Kaur, an epidemiologist and infectious-disease specialist at St. Christopher’s Hospital for Children in Philadelphia. The reduced ACE2 gene expression in children suggests, but doesn’t prove, that children create less of the protein that the virus uses as its gateway, or receptor. “It’s a signal, but does that translate to lower protein production, or less receptor activity?” Kaur asked. “Like any good study, it generates its own set of questions.” » HELP US REPORT: Are you a health care worker, medical provider, government worker, patient, frontline worker or other expert? We want to hear from you.  ACE2 (short for angiotensin converting enzyme 2) is part of a complex system that regulates blood pressure, fluid, and mineral balance in the body. Because ACE2 also helps the coronavirus take hold in the nose, lungs, and other organs, it has become a subject of interest. For example, researchers are studying whether common blood pressure medicines that stimulate ACE2 can make COVID-19 more deadly — or less so because ACE2 tamps down inflammation and tissue scarring. The mystery of COVID-19 in children has been as puzzling as it is a relief. Children, who make up fewer than 2% of identified cases, generally have mild symptoms or none, and rarely die of the infection. That’s the opposite of the seasonal influenza, which can be devastating in young children because their immune systems have not matured, and they don’t have prior exposure to most flu strains. Flu vaccination is recommended to protect them. To see whether the ACE2 gene may play a role, the Mount Sinai team used nasal lining samples that were provided by 305 people for a study conducted between 2015 and 2018. Half of them had asthma, which was the focus of the study. The samples were categorized by age — children under 10, children 10 to 17, young adults 18 to 24, and adults 25 and over. ACE2 gene activity was lowest in children under 10, and rose steadily and significantly with age. (The results were statistically adjusted to make sure asthma didn’t skew them.) Although the researchers, led by pediatrician Supinda Bunyavanich, found ACE2 gene expression was linked to age, they noted that a study conducted before the pandemic of patients with severe respiratory distress found no link between ACE2 protein activity and age. However, that study didn’t examine gene expression, and “the lung and nasal environments are distinct, with known differences in gene expression." |
|
|
|
Post by Admin on May 24, 2020 19:27:28 GMT
Nasal Gene Expression of Angiotensin-Converting Enzyme 2 in Children and Adults
Supinda Bunyavanich, MD, MPH1; Anh Do, PhD2; Alfin Vicencio, MD1
JAMA. Published online May 20, 2020. doi:10.1001/jama.2020.8707
Comment
Children account for less than 2% of identified cases of coronavirus disease 2019 (COVID-19).1,2 It is hypothesized that the lower risk among children is due to differential expression of angiotensin-converting enzyme 2 (ACE2),3 the receptor that severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) uses for host entry.4 We investigated ACE2 gene expression in the nasal epithelium of children and adults.
Methods
We conducted a retrospective examination of nasal epithelium from individuals aged 4 to 60 years encountered within the Mount Sinai Health System, New York, New York, during 2015-2018. Samples were collected from individuals with and without asthma for research on nasal biomarkers of asthma. The study was approved by the Mount Sinai institutional review board. Written informed consent was obtained from participants (or their parents for minors). Nasal epithelium was collected using a cytology brush that was immediately placed in RNA stabilization fluid and stored at −80 °C. RNA was isolated within 6 months. RNA samples were checked for quality and sequenced as a single batch in 2018. Sequence data processing included sequence alignment and normalization of gene expression counts across genes and samples.
Given the role of ACE2 in SARS-CoV-2 host entry,4 ACE2 gene expression was the focus of this study. Linear regression models with and without adjustment for covariates (sex and asthma) were built with ACE2 gene expression in log2 counts per million as the dependent variable and age group as the independent variable using R software, version 3.6.0 (R Foundation). Age was categorized into the following groups reflecting developmental life stages: younger children (aged <10 years), older children (aged 10-17 years), young adults (aged 18-24 years), and adults (aged ≥25 years). Two-sided tests and a significance threshold of P ≤ .05 were used. Trend pattern was evaluated using polynomial orthogonal contrasts.
Results
The cohort of 305 individuals aged 4 to 60 years was balanced with regard to sex (48.9% male). Because the cohort had been recruited to study biomarkers of asthma, 49.8% had asthma.
We found age-dependent ACE2 gene expression in nasal epithelium (Figure). ACE2 gene expression was lowest (mean log2 counts per million, 2.40; 95% CI, 2.07-2.72) in younger children (n = 45) and increased with age, with mean log2 counts per million of 2.77 (95% CI, 2.64-2.90) for older children (n = 185), 3.02 (95% CI, 2.78-3.26) for young adults (n = 46), and 3.09 (95% CI, 2.83-3.35) for adults (n = 29).
Linear regression with ACE2 gene expression as the dependent variable and age group as the independent variable showed that compared with younger children, ACE2 gene expression was significantly higher in older children (P = .01), young adults (P < .001), and adults (P = .001) (Figure). As the distributions of sex and asthma varied among the age groups, a linear regression model adjusted for sex and asthma was built that also showed significant adjusted associations (P ≤ .05) between ACE2 expression and age group. Regression (β) coefficients for age groups from the unadjusted and adjusted models are shown in the Table. These regression coefficients indicate the difference in ACE2 expression (in log2 counts per million) between a given age group and the group of children younger than 10 years. Tests for trend using polynomial orthogonal contrasts indicated a significant linear trend for change in ACE2 expression with advancing age group (P ≤ .05).
Discussion
The results from this study show age-dependent expression of ACE2 in nasal epithelium, the first point of contact for SARS-CoV-2 and the human body. Covariate-adjusted models showed that the positive association between ACE2 gene expression and age was independent of sex and asthma. Lower ACE2 expression in children relative to adults may help explain why COVID-19 is less prevalent in children.3 A limitation of this study is that the sample did not include individuals older than 60 years.
Few studies have examined the relationship between ACE2 in the airway and age. A study of bronchoalveolar lavage fluid from 92 patients with acute respiratory distress syndrome reported no association between ACE2 protein activity and age,5 but epithelial gene expression was not examined, and ACE2 protein may be variably shed into bronchoalveolar lavage fluid. Furthermore, the lung and nasal environments are distinct, with known differences in gene expression.6 This study provides novel results on ACE2 gene expression in nasal epithelium and its relationship with age.
|
|
|
|
Post by Admin on May 25, 2020 8:10:32 GMT
Mechanism of SARS-CoV-2 infection On binding to ACE2, the spike protein undergoes host cell proteolytic cleavage into two subunits: S1, which contains the receptor-binding domain (RBD) and S2, which enables fusion with the host cell membrane and viral entry. “A cell-surface host serine protease, TMPRSS2 [transmembrane serine proteinase 2], is also thought to be involved in viral entry and is proposed to cleave S1 and S2, leading to activation of the fusion machinery,” write Peter Monk and colleagues. The new assay used cells that express both ACE2 and TMPRSS2 To investigate SARS-CoV-2 binding to host cells, the team developed a new assay using the RT4 urinary bladder transitional carcinoma cell line, which expresses both ACE2 and TMPRSS2. They found that an intact recombinant form of the viral spike protein containing both S1 and S2 (S1S2), but not the S1 domain alone, binds strongly to RT4 cells in a temperature-dependent manner. Binding activity sharply increased at 37°C, suggesting that proteolytic cleavage was likely to be involved, says the team. .jpg&ts=20200524085300&ri=674) Are there any other mechanisms of viral entry? Monk and colleagues say that most cell types only express quite low levels of ACE2, suggesting that the spike protein might also interact with other receptor sites to gain viral entry. Certain viruses such as herpes simplex are already known to bind with host glycosaminoglycans called heparan sulfates, says the team. In addition, a study by one group suggested that the soluble glycosaminoglycan heparin can inhibit the entry of SARS CoV-2 into “Vero” cells – a cell line derived from monkey kidney epithelia. “These authors also showed that heparin could interact with recombinant S1 RBD and cause conformational changes, leading to the suggestion that SARS-CoV-2 might use host heparan sulfates as an additional attachment site during infection,” write the researchers. Unfractionated heparin completely stopped the binding Given that the new assay already seemed to mimic some features of SARS-CoV-2 infection, the researchers used it to test the effects of incubating RT4 cells with heparin at 37°C. The team reports that unfractionated heparin (UFH) completely inhibited the binding of S1S2 to RT4 cells. Treating the cells with two low molecular weight heparins (LMWHs) that are already in clinical use also inhibited the binding, but only partially and not as strongly. “This suggests that heparin, particularly unfractionated forms, could be considered to reduce clinical manifestations of COVID-19 by inhibiting continuing viral infection,” write Monk and team. Could the spike protein also bind host cell heparan sulfate? The authors say the interaction they observed between heparin and the spike protein suggests that it might also bind to host cell heparan sulfate. To test this hypothesis, they treated RT4 cells with a blend of heparinase I and III, enzymes that degrade heparan sulfate molecules, before testing the binding of S1S2. The treatment did not result in any significant reduction in the binding of RT4 cells, suggesting that heparan sulfates do not play any significant role in the attachment of SARS-CoV-2 spike protein to host cells: “Although our data supports the inhibitory activity of UFH, it does not support the conjecture that heparan sulfates are essential for viral infection,” writes the team. What are the implications of the study? The researchers say that LMWHs, which have already been used to treat COVID-19 patients and been shown to improve outcomes, are much smaller than UFH and have pharmacokinetics that is easier to predict. Monk and colleagues think their work suggests that earlier use of heparin should be considered when a viral infection is still an important factor in influencing the severity of disease. “The use of UFH rather than LMWH should also be considered, although we note that administration and the safety profile of UFH might preclude this in some cases,” they add. Finally, the researchers say their newly developed flow cytometric assay for assessing the binding of SARS-CoV-2 spike protein to host cells lends support to a previous finding that heparin can inhibit viral attachment to monkey kidney epithelial cells. “Our new assay could be a useful first screen for novel inhibitors of coronavirus infection,” concludes the team. *Important Notice bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information. Journal reference: Monk P, et al. Unfractionated heparin potently inhibits the binding of SARS-CoV-2 spike protein to a human cell line. bioRxiv 2020. doi: doi.org/10.1101/2020.05.21.107870 |
|
|
|
Post by Admin on Oct 23, 2020 19:39:13 GMT
"The starting point of our study was the question why SARS-CoV, a coronavirus that led to a much smaller outbreak in 2003, and SARS-CoV-2, spread in such a different way even if they use the same main receptor ACE2", says University of Helsinki virologist Ravi Ojha.
A crucial piece of the puzzle appeared on comparing the two viral genomes; SARS-CoV-2 had picked up sequences responsible for producing a prickly array of 'hooks', not unlike those used by other nasty pathogens to grip onto host tissues.
"Compared to its older relative, the new coronavirus had acquired an 'extra piece' on its surface proteins, which is also found in the spikes of many devastating human viruses, including Ebola, HIV, and highly pathogenic strains of avian influenza, among others," says Olli Vapalahti, also a virologist from the University of Helsinki.
"We thought this could lead us to the answer. But how?"
Consulting with colleagues around the world, the researchers zeroed in on neuropilin-1 as a common factor.
Typically, this receptor plays a role in responding to growth factors important in tissue development, especially among nerves. But to many viruses, it's a convenient handle for holding onto host cells long enough to break in.
Electron microscopy of the surface spikes coating SARS-CoV-2 particles certainly hinted at the potential for a relationship with the receptor.
To help confirm it, the researchers made use of monoclonal antibodies specifically selected to block access to garden variety neuropilin-1, but not to mutant varieties tweaked to have a slightly different structure.
Sure enough, 'pseudoviruses' sporting SARS-CoV-2 proteins (great for watching viruses enter cells without worrying about the whole messy replication business that follows) had a much harder time getting inside when neuropilin-1 was locked up.
"If you think of ACE2 as a door lock to enter the cell, then neuropilin-1 could be a factor that directs the virus to the door," says Balistreri.
"ACE2 is expressed at very low levels in most cells. Thus, it is not easy for the virus to find doors to enter. Other factors such as neuropilin-1 might help the virus finding its door."
With neuropilin-1 expressed in large amounts in nerve tissues within the nasal cavity, we might imagine SARS-CoV-2 has a convenient red carpet rolled out for it the moment we sniff an infected droplet.
A close look at tissue samples expressing neuropilin-1 taken from deceased COVID-19 patients added to suspicions, while an experiment involving mice helped confirm the receptor's role in assisting the virus's entry into our nervous system.
Whether this might help explain why SARS-CoV-2 infections can have such a traumatic impact on the brain's function is a question for future research.
"We could determine that neuropilin-1, at least under the conditions of our experiments, promotes transport into the brain, but we cannot make any conclusion whether this is also true for SARS-CoV-2. It is very likely that this pathway is suppressed by the immune system in most patients," says neurologist Mika Simons from the Technical University of Munich.
It's tempting to picture new forms of antiviral medication on the horizon. Though as rapidly as SARS-CoV-2 reveals its criminal talents, simply blocking off cell receptors is likely to be bad news for our health.
That's not to say the discovery isn't without opportunity.
"Currently our laboratory is testing the effect of new molecules that we have specifically designed to interrupt the connection between the virus and neuropilin," says Balistreri.
"Preliminary results are very promising and we hope to obtain validations in vivo in the near future."
This research was published in Science.
Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity
Abstract
The causative agent of coronavirus induced disease 2019 (COVID-19) is the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). For many viruses, tissue tropism is determined by the availability of virus receptors and entry cofactors on the surface of host cells. Here, we found that neuropilin-1 (NRP1), known to bind furin-cleaved substrates, significantly potentiates SARS-CoV-2 infectivity, an effect blocked by a monoclonal blocking antibody against NRP1. A SARS-CoV-2 mutant with an altered furin cleavage site did not depend on NRP1 for infectivity. Pathological analysis of human COVID-19 autopsies revealed SARS-CoV-2 infected cells including olfactory neuronal cells facing the nasal cavity positive for NRP1. Our data provide insight into SARS-CoV-2 cell infectivity and define a potential target for antiviral intervention.
|
|
|
|
Post by Admin on Oct 24, 2020 19:47:22 GMT
An outbreak of SARS-CoV-2 infections has caused a pandemic associated with a severe acute pulmonary disease named COVID-19 (coronavirus induced disease 2019) (1). A related coronavirus, SARS-CoV, led to a much smaller outbreak in 2003, possibly due to infection occurring predominantly in the lower respiratory system, whereas SARS-CoV-2 spreads rapidly through active pharyngeal viral shedding (2). Despite these differences, uptake of both viruses is mediated by the identical cellular receptor, angiotensin-converting enzyme 2 (ACE2) (3–5). One attractive hypothesis to explain the enhanced spreading of SARS-CoV-2 is the presence of a polybasic furin-type cleavage site, RRAR^S, at the S1/S2 junction in the SARS-CoV-2 spike (S) protein that is absent in SARS-CoV (6). Similar sequences are found in the S proteins of many other pathogenic human viruses, including Ebola, HIV-1 and highly virulent strains of avian influenza (6, 7). The presence of the polybasic cleavage site in SARS-CoV-2 results in enhanced pathogenicity by priming the fusion activity (8) and could potentially create additional cell surface receptor binding sites. Proteolytic cleavage of RRAR^S by furin exposes a conserved carboxyterminal (C-terminal) motif RXXROH (where R is arginine and X is any amino acid; R can be substituted by lysine, K) in the S protein. Such C-terminal sequences that conform to the “C-end rule” (CendR) are known to bind to and activate neuropilin (NRP1 and NRP2) receptors at the cell surface (9). Recent cryo-electron microscopy structures of the SARS-CoV-2 S protein demonstrated that the S1/S2 junction is part of a solvent-exposed loop and therefore accessible for receptor interactions (10, 11). To determine whether SARS-CoV-2 can use NRP1 for virus entry and infectivity, we generated lentiviral particles pseudotyped with the SARS-CoV-2 S protein. Pseudoviruses are well suited for virus entry assays, as they allow viral entry to be distinguished from other virus life cycle steps. HEK-293T cells, which have almost no detectable ACE2 and NRP1 transcripts (fig. S1), were transfected with plasmids encoding the two established host factors (4), human ACE2 and the transmembrane protease serine 2 (TMPRSS2), or NRP1. When expressed alone, ACE2 rendered cells susceptible to infection (Fig. 1A). While NRP1 only hardly promoted infection in HEK-293T cells, its co-expression with ACE2 and TMPRSS2 markedly enhanced infection (Fig. 1, A and B). NRP1 expression increased infection in Caco-2 cells, which endogenously express ACE2 (12) (Fig. 1C and fig. S1D), showing that NRP1 can potentiate infection in the presence of other host factors. To test the specificity of NRP1-dependent virus entry, we developed monoclonal antibodies (mAbs) that were designed to functionally block the extracellular b1b2 domain of NRP1, known to mediate the binding to CendR peptides (13). The mAb3 was observed to bind to the recombinant b1b2 domain of wild-type NRP1, but not to the triple mutant b1b2 domain (S346A, E348A and T349A in the CendR binding pocket) (fig. S2A). The potency of the mAbs in preventing cellular binding and internalization of NRP ligands was tested using 80 nm silver nanoparticles (AgNP) coated with the prototypic NRP1-binding CendR peptide RPARPAROH (9) (fig. S2B). mAb3 efficiently blocked AgNP-CendR binding (fig. S2C) and internalization (fig. S2, D and E), while another monoclonal antibody, mAb2, had no effect and was used as a control in further experiments. Treatment of HEK-293T with mAb3 significantly reduced infection by SARS-CoV-2 pseudoviruses in cells expressing ACE2/TMPRSS2/NRP1 (Fig. 1D), but not in cells expressing ACE2/TMPRSS2 only (fig. S2F). When SARS-CoV-2 pseudovirus was pre-incubated with recombinant, soluble extracellular b1b2 domain of NRP1, the wild-type significantly reduced infection compared to the triple mutant (Fig. 1E and fig. S2G).  Fig. 1 NRP1 facilitates the cellular entry of SARS-CoV-2 pseudotyped particles. (A) Representative images and quantification of SARS-CoV-2 spike protein (SARS-2) (blue bars) and VSV-G pseudotype (gray bars) infectivity in HEK-293T cells transiently expressing control (ctrl) vector, ACE2, NRP1 or TMPRSS2 (TSS2). Data are normalized to the respective infectivity of SARS-2 and VSV-G pseudotype in ACE2-expressing cells. Two-way ANOVA with Tukey’s correction for multiple comparisons. (B) HEK-293T cells transiently expressing ACE2 and TMPRSS2 or NRP1, ACE2 and TMPRSS2 were inoculated with SARS-2 pseudotype. Data are normalized to SARS-2 infectivity in cells expressing ACE2 and TMPRSS2. One-way ANOVA with Tukey’s correction for multiple comparisons. (C) SARS-2 pseudotype infectivity in Caco-2 cells expressing NRP1 or control vector. Data are normalized to the respective infectivity of SARS-2 and VSV-G pseudotype in control cells. Two-way ANOVA with Sidak’s correction for multiple comparisons. (D and E) HEK-293T cells transiently expressing NRP1, ACE2 and TMPRSS2 were inoculated with SARS-2 pseudotype in the presence of mAb3 antibody against NRP1 (D, mAb3) or control mAb2 (D, ctrl Ab), and in the presence of soluble NRP1 wild-type b1b2 domain (E, wt b1b2) or the NRP1 mutant b1b2 domain (E, mut b1b2). Data in (E) are normalized to untreated cells expressing NRP1, ACE2 and TMPRSS2. Two-tailed unpaired Student’s t test. All data are represented as mean ± s.d. from three independent experiments (A to C) or three biological replicates (D and E). *p < 0.05, **p < 0.01, ****p < 0.0001. All images show GFP-positive, infected cells (magenta) and Hoechst (cyan). Scale bars, 100 μm. Next, we explored the role of NRP1 using SARS-CoV-2 isolated from COVID-19 patients from the Helsinki University Hospital. We used wild-type SARS-CoV-2 and a cleavage-impaired SARS-CoV-2 mutant that was isolated from Vero-E6 which rapidly accumulate mutations at the furin cleavage site of the S protein over passaging (Fig. 2, A and B) (14). First, we confirmed that furin cleaved the wild-type, but not the mutant, SARS-CoV-2 S protein by analyzing S protein processing in CHO cells with functional (parental) or deficient (FD11) furin enzyme (fig. S3) (15). Next, we validated that exogenous ACE2 expression rendered HEK-293T cells susceptible to infection with SARS-CoV-2 (Fig. 2, C and D). NRP1 expression alone caused lower levels of infection, which were only detectable with increasing virus titer (Fig. 2, C and D). We then compared the ability of wild-type and mutant SARS-CoV-2 virus to infect HEK-293T stably expressing either ACE2, ACE2/TMPRSS2 or ACE2/TMPRSS2/NRP1. Infection of these cell lines by the wild-type, but not the mutant, virus increased in the presence of NRP1, providing further evidence that NRP1 requires a furin-cleaved substrate for its effects (Fig. 2, E and F). Next, we studied the effect of the NRP1-blocking antibody, mAb3, on infection of Caco-2 cells by wild-type and mutant SARS-CoV-2 virus, and found that pre-incubation with NRP1-blocking antibody reduced wild-type virus infection by ~40%, while the control mAb2 had no effect (Fig. 2, G and H). NRP1-blocking antibody had no effect on the infection by the mutated virus (Fig. 2, G and H).  Fig. 2 A blocking antibody against the b1b2 domain of NRP1 reduces infection by wild-type SARS-CoV-2 (SARS-2-wt), but not a mutant with a deletion at the furin-cleavage site (SARS-2-mut). (A) Sequence analysis of viruses isolated at different passages (P) from different cell types. The first sequence is the reference from the Wuhan isolate (NC_045512.2). The sequence abundance in each virus population is indicated as a percentage (%). § = SARS-2-wt, §§ = SARS-2-mut, used in the experiments. (B) A deletion adjacent to the furin-cleavage site abrogates the enzymatic cleavage of the S protein. Immunoblot analysis of cell lysates from Vero-E6 cells infected for 16h with two viral populations (§ and §§). (C and D) Representative images and quantification of SARS-2-wt infectivity in HEK-293T cells stably expressing ACE2 (blue bars) or NRP1 (orange bars) compared to non-transfected cells (gray bars). Different virus titers were used. Data are normalized to the infectivity in ACE2-expressing cells at MOI 5. Two-way ANOVA with Tukey’s correction for multiple comparisons. (E and F) Representative images (E) and quantification (F) of HEK-293T cells stably expressing the indicated combinations of ACE2, TMPRSS2 (TSS2) and NRP1 following SARS-2-wt (wt, blue bars) or SARS-2-mut (mut, gray bars) inoculation. Data are normalized to the respective infectivity in ACE2-expressing cells. Two-way ANOVA with Tukey’s correction for multiple comparisons. (G and H) Caco-2 cell infection in the presence of control mAb2 (ctrl. Ab) or mAb3 blocking antibodies against NRP1 after SARS-2-wt (wt, blue bars) or SARS-2-mut (mut, gray bars) inoculation. Data are normalized to the respective vehicle control (PBS) sample. Two-way ANOVA with Tukey’s correction for multiple comparisons. Data are mean ± s.d. from three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. SARS-2-wt and SARS-2-mut infected cells (magenta), Hoechst (cyan). Scale bars, 50 μm. Cleavage of SARS-CoV-2 S protein at the S1/S2 site generates the C-terminal end TQTNSPRRAROH. To determine whether this specific sequence can function as a substrate for NRP1, we used AgNPs coated with TQTNSPRRAROH peptide or different control peptides including one with a terminal amide group, which reduces NRP1 binding (TQTNSPRRARNH2) (9) (Fig. 3A). We found that AgNP-TQTNSPRRAROH, but not control AgNPs were efficiently taken up by HEK-293T cells expressing NRP1 (Fig. 3, B and C). Next, we determined whether AgNP-TQTNSPRRAROH particles were also internalized into cells in vivo. We chose to study nanoparticle entry in the mouse olfactory epithelium, owing to the known expression of NRP1 in the olfactory system (16) including olfactory neuronal cells of the epithelium (fig. S4). AgNPs-TQTNSPRRAROH and control AgNP-TQTNSPRRARNH2 were administered into the nose of anesthetized adult mice. 6 hours after administration, we observed a significantly larger uptake of AgNP-TQTNSPRRAROH compared to AgNP-TQTNSPRRARNH2 into the olfactory epithelium (Fig. 3, D and E), and unexpectedly also into neurons and blood vessels of the cortex (Fig. 3, F and G). Similar results were obtained for AgNPs coated with the prototypic NRP1-binding CendR peptide RPARPAROH (fig. S5). |
|

